|
Artículos de Revisión
Genetic of obesity
Eduardo Cuestas

Revista Facultad de Ciencias Medicas 2008; 65(4): 117-124
Concept, acquisition,
interpretation of data, drafting the article and writing the
final version.
Department of Pediatrics and Neonatology. Hospital Privado
Centro Médico de Córdoba. Academic Unit. School of Medicine.
National University of Cordoba. Argentina.
Prof. Dr. E Cuestas. Servicio de Pediatría y Neonatología.
Hospital Privado. Naciones Unidas 346
Córdoba X50016KEH. Telephone: 54-351-4688241. Fax:
54-351-4688286 e-mail.
Obesity has become a major health problem around the world,
with an increasing incidence in children[1] . The genetic
study of obesity is progressing with high velocity. This
review discusses the relationships between genetics and
obesity, and the role of genetics in potential obesity
treatment.
Obesity is defined as an excess of body fat. In the practice,
physicians use a body mass index (BMI = body weight in
Kilograms divided by square of height in meters) > 30 to
define obesity. BMI charts are now available for children by
age and sex [2] .
In the last 20 years has yielded the tools necessary to
explore the biochemistry underlying obesity, it has also
demonstrated that interactions between genetic and
environment are critical for the regulation of adipose mass
function. Medical and nutritional recommendations based on
genetically undefined and/or environmentally heterogeneous
population-based studies have, not unsurprisingly, minimal
success in treating common obesity. It is precisely this
lack of success that is conducting for the discussed
concepts of personalized medicine[3,4] and nutrition[5-7].
However, prior realizing these concepts, the genetic
components underlying common obesity must be elucidated.
In the later ten years, the study of genetically complex
diseases has benefited from the advances in molecular
biology. While obesity was understand how a problem that
obeys rules of genetic laws, new technologies have led to
unsuspected developments. Obesity originated from a single
dysfunctional gene (i.e., monogenic obesity) is both severe
and rare when compared to the more common form of obesity,
in which numerous genes make minor contributions in
determining phenotype (i.e., polygenic obesities). Some
genetic candidates underlying monogenic obesities in the
mouse have been defined, transferring this knowledge to
humans has led to more questions than answers. However,
molecular approach has revealed novel candidate genes for
the various types of human obesity, has suggested that
several clinical cases previously defined as monogenic
obesity are genetically more complex, and has show genetic
and environmental interactions as fundamentally important to
understanding the mechanisms involved in fat-mass
pathophysiology.
However, a genetic approach may provide a useful framework
for addressing the obesity epidemic, more research is needed
before specific targeted health interventions can be
effectively integrated in the clinical practice.
Genetics and environmental factors
Energy storage in the form of fat is a very important
adaptation to survival. It is probable that a group of genes
have been selected during evolution to favor energy storage
(“thrifty gene hypothesis” [8] ). In an actual context of
increased food availability and decreased physical activity,
these genes will confer susceptibility to develop obesity
and its perpetuation[9]. The role of genes in body weight
control is based on studies in monozygotic twins who show a
high concordance in body composition and response to
overfeeding[9] . The susceptibility in most cases is
polygenic, and rarely monogenic.
Moreover, genetic factors can modulate nutrient storage, but
also, nutrients modulate gene expression too[10]. Obesity
results from very complex genetic and environmental
interactions.
Interactions
Genomics is the study of the entire human genome and
involves not only studying the actions of single genes but
also the interactions of multiple genes with each other and
with the environment. This review emphasizes the
multifactorial nature of obesity, which is caused by the
interaction of genes, environment, and lifestyle.
Separating genetic factors from other factors is difficult
because genes are part of dynamic system that is constantly
in flux in response to environmental cues, for this reason
at the present is not sufficiently clear the causal relation
between genes and body weight regulation.
Obesity is a complex disorder with contribution of multiple
genes and gene variants. Evidence suggests that genetics
factors are involved in all aspects of weight regulation,
including food intake, energy expenditure, hunger, appetite,
eating behavior, taste, satiety, spontaneous physical
activity, metabolic rate, thermogenesis, and motivation to
exercise. Same genetic research gives a sight of the complex
relations between genes, genes and age, and genes and
environment. Genome wide association studies have led to
rapid progress in the understanding of genetic basis of
diseases and specially in genetics interactions.
A study[11] found a significant interaction between two gene
variants, PPAR λ 2 and ADR β 3, in the risk of obesity in
children and adolescents. After adjusting for family history
of obesity, the researchers found that carriers of both gene
variants were almost 20 times more likely to be obese than
noncarriers, suggesting a synergistic effect between the two
genes.
The effects of genes may vary depending on age. Investigator[12]
reported an association between a mutation in the gene for
agouti-related protein (a strong appetite stimulator) and
obesity in older adults. The mutation was not associated
with obesity in study subjects with a mean age of < 25 years,
but was significantly associated with fat and abdominal
adiposity in parents.
A great number of studies explored gene–environment–lifestyle
interactions. Researchers[13] found that an interaction
between diet and specific genes may affect obesity. Exist an
increased obesity risk for women with a Glu27 variant and a
diet with more than 49% of calories coming from
carbohydrates. An alternate variant of that same gene was
not associated with an increased obesity risk in relation to
carbohydrate intake, given the same number of calories
consumed. This finding suggests that the effect of diet on
obesity depends on genetic factors. Changes in gene
expression that result from epigenetic influences (modifications
of DNA structure rather than DNA sequence) were also
explored because of their potential role in obesity and
associated chronic diseases. Associations between fetal
environment (Barker theory) and adult-onset obesity may be
due to epigenetic influences that promote fat storage, but
possible mechanisms are not well understood[14,15].
Genes, metabolic disorders and secondary obesity
Distribution of fat, more than amount of total body fat,
play a key role in metabolic consequences associated with
obesity. Excess central (abdominal) fat is particularly
associated with adverse effects. People with fat
concentrated in the abdomen are more likely to develop
diabetes than are people with the same amount of fat
distributed throughout the body. A study in prepubertal
children also suggests that genetics has a role in abdominal
fat deposition[16].
There are near 30 genetics diseases in which patients are
obese, with different grades of mental abnormalities,
dysmorphic features, and organ-specific developmental
dysfunction[17,18]. Such cases are called syndromic obesity.
These syndromes arise from genetic defects or chromosomal
abnormalities, and can be either autosomal or X-linked. The
genetic component analysis suggests that multiple genes
within a biological pathway may produce identical phenotypes
[19] . The most common disorders known are Prader-Willi
syndrome (PWS), Bardet-Biedl syndrome (BBS), and Alström
syndrome, but many others have been reported too [19] .
The most frequent of these syndromes (1 in 25,000 births) is
PWS, which is characterized by obesity, hyperphagia,
diminished fetal activity, mental retardation, and
hypogonadism. This disease is caused by an absence in the
paternal segment 15q11.2–q12 through chromosomal loss.
Several candidate genes in the 15q11–13 region of PWS
patients have been studied; however, the molecular basis of
hyperphagia remains undefined in part because of the fact
that none of the currently available PWS mouse models have
an obese phenotype[20]. One candidate protein that may
mediate the severe hyperphagia of PWS is the gastric hormone
ghrelin[21], via its regulation of hunger and stimulation of
growth hormone [18] . Ghrelin's role is further implied by
the positive findings that growth hormone supplementation is
capable of reversing several dysfunctional processes
associated with PWS[22-23]; however, in the absence of a
suitable experimental model, identifying the genetic
components of this syndrome will be difficult.
BBS is characterized by obesity, rod-cone dystrophy,
morphological finger abnormalities, learning difficulties,
and renal disease, among other clinical manifestations. It
was originally classified as a homogeneous syndrome, in the
present BBS has been associated with at least 11 different
chromosomal locations, with several mutations identified
within some of the following locations: BBS1 on 11q13, BBS2
on 16q21, BBS3 on 3p13, BBS4 on 15q22.3, BBS5 on 2q31, BBS6
on 20p12, BBS7 on 4q27, BBS8 on 14q32.11, BBS9 on 7p14,
BBS10 on 12q21.2, and BBS11 on 9q33.1 [24-27]. While BBS was
considered to be autosomal-recessive, it has recently been
found that the clinical symptoms of certain forms of BBS are
related to recessive mutations at one of the BBS loci
associated with a heterozygous mutation at a second locus,
prompting, for the first time, the hypothesis of a
triallelic mode of transmission[28]. Six genes are
characterized in BBS. For the BBS6 locus, positional cloning
identified the MKKS gene, which codes for a chaperone
protein. Mutations identified in MKKS result in a shortened
chaperone protein and are present in 5%–7% of BBS cases;
however, the links between MKKS, its eventual target
proteins, and the BBS clinical traits are largely unknown. A
newly identified locus, BBS10, has recently been found to
code for C12orf58, a vertebrate-specific chaperone-like
protein, and was found to be mutated in 20% of the
populations examined from various ethnic backgrounds [25].
Unlike BBS6 and BBS10, the genes associated with BBS1, BBS2,
and BBS4 are very different from MKKS and C12orf58 genes,
but it is reasonable that they code for protein substrates
of these chaperones [29]. Recently, the gene encoding the E3
ubiquitin ligase TRIM32 was identified as the 11th locus
associated with BBS, suggesting that the list of genetic
components for this syndrome may yet remain incomplete [27].
Functional works performed in single-cell organisms have
shown that certain BBS genes are specific to ciliated cells[30].
Ciliated cells have a role in mammalian development,
contributing to right/left asymmetry, thus enabling the
organs to be correctly positioned within the biological
system. Dysfunction in processes affecting ciliated cells
may contribute to the alterations in pigmentary epithelia
and structural anomalies noted in certain organs in patients
with BBS; however, the relationship between cilia and
obesity remains unclear [31]. Recent findings suggest that
cilia formation is not dependent on BBS gene functionality
[32]; rather, the BBS genes may play an important role in
intracellular signaling [31]. An article used Caenorhabditis
elegans to demonstrate the regulation of fat storage by
orthologs of both neuronal TUB and BBS1 genes, suggesting
the existence of a currently unidentified intertissue
signaling pathway that may link ciliated neurons and adipose
cells[33,34].
While syndromic obesity was previously presumed to be under
the control of a single gene, progress in genomic era has
clearly distinguished this type of obesity, defining the
contribution of multiple genetic factors in a syndrome is
significantly more challenging than localizing the single
gene involved in monogenic diseases.
Monogenic Obesity
Nearly 200 cases of human obesity have been associated with
a single gene mutation. These cases, which obey Mendelian
genetics, are characterized by severe phenotypes that
present themselves in childhood and are often associated
with additional behavioral, developmental, and endocrine
disorders
The first gene identification was made in animal models of
monogenic obesity. These rodents models mostly involved
genes in regulatory pathway of food intake. Later, similar
affected ways have been discovered in human monogenic
obesity[35].
The present model have two integrated pathways: a leptin,
leptin receptor (LR) and a melanocortin pathway. Leptin is
produced by adipocytes . It bind to hypothalamic receptors
in the arcuate nucleus and this induced an increased
secretion and syntesis of α-melanocyte stimulating hormone
(α-MSH) that is formed from pro-opiomelanocortin (POMC)
through proteolitic cleavage mediated by pro-hormone
convertase 1 (PC-1) . α-MSH binds to the melanocortin 4
receptor (MC4R) in the paraventricular nucleus. This in turn
inhibit the effectors of food intake (see fig. 1).
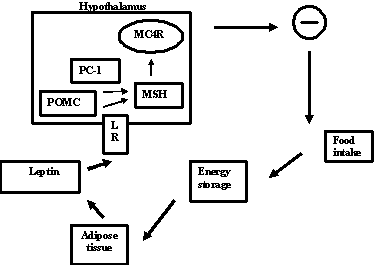
Fig 1.
Schematic representation of leptin regulation
food intake in monogenic obesity. Leptin is secreted by
adopocytes. It bind to hypothalamic receptor (LR) and this
induces an increased synthesis of melanocyte stimulating
hormone (MSH), formed from pro-opiomelanocortin (POMC)
through proteolytic cleavage mediated by pro-hormone
convesrase 1 (PC-1). MSH binds to the melanocortin 4
receptor (MC4R). This, in turn, inhibit the effectors of
food intake (-).
Homozygous carriers of loss or mutation in the leptin gene
exhibit early obesity onset, hypogonadotrophic hypogonadism
and central hypohyroidism. The treatment with recombinant
human leptin is in this case looks promising[35 ].
The homozygous mutation of leptin receptor, abolish leptin
signaling, leading to a similar picture that individuals
with leptin deficiency , but also display growth retardation.
Because α-MSH is involved in the regulation of food intake
and also in hair pigmentation, defects in POMC function
include obesity, red hair and ACTH deficiency.
Mutations of PC-1 have obesity, ACTH deficiency and
hyperproinsulinemia, because PC-1 is involved in the
conversion of proinsulin into insulin in pancreatic β-cells.
Mutations in MC4R cause dominant an recessive inherited
nonsyndromic obesity, with incomplete penetrance and
variable expression. It is similar to more common forms of
obesity, with a early age onset. MC4R mutations represents a
important cause of obesity between children (1-6%)[36].
Obesity in which a single gene can be can be identified as
the major cause of disease is rarely found.
Polygenetic obesity
Polygenic or common obesity arises when an individual
genetic profile develop in an environment that promotes
energy consumption over energy expenditure. Every time more
societies have an environment that favors weight gain rather
than loss because of food abundance and lack of physical
activity, thus positioning common obesity as a major
epidemic currently challenging these societies. Many very
good reviews have been published in which the genetic
complexity and the challenges in dissecting the perturbed
biology underlying common obesity have been outlined
[17,37,38-40]. While independent replication of a novel
association is mandatory, it is important to stress that our
current degree of understanding of genetics and environment
(G&E) interactions should prevent an unreplicated result
from immediately being discarded. Complex traits are highly
dependent on G&E interactions; however, a question persists:
how can one account for and control all possible influences
within an experimental design that may affect data
interpretation and ultimately experimental conclusions? The
individual complexity in humans relates to the alleles that
influence common diseases in different genetic ways, and to
variable genetic combinations eventually influenced by
different epigenetic (including in utero) or environmental
factors during an individual's lifetime (the role of
epigenetics has been thoroughly reviewed in[41,42]). The
precise degree to which genes eventually contribute to
complex traits remains poorly defined, and the importance of
subtle environmental factors may simply not be appreciated.
Studies of polygenic obesity are based on the analysis of
single nucleotide polymorphisms (SNPs) or repetition of
bases (polyCAs or microsatellites) located within or near a
candidate gene, where a candidate gene is one that meets a
number of criteria, such as its proximity to a quantitative
trait locus or its having a phenotypic effect following
genetic manipulation (e.g., knock-out and knock-in models)
[43]. Should a candidate-gene variant appear promising based
on results derived from in vitro and animal-model studies,
its association with the obese phenotype is then examined in
case-control and family studies[44]; however, many genes and
chromosomal regions contribute to defining the common obese
phenotype [45,46]. These genes have been implicated in a
wide variety of biological functions, such as energy
expenditure, the regulation of food intake, glucose and
lipid metabolism, and adipose tissue development.
Not only the number of genes associated with obesity is high,
but variants in some of these genes are demonstrating the
importance of polymorphisms in the “interpretation” of
environmental stimuli [5]. In contrast to animal models
whose environments can be rigorously controlled, the genetic
and environmental diversity in humans has proved problematic
for data replication [44,45,47] and the result can be
conflicting, for example, an association between three SNPs
of GAD2, which codes for the 65-kDa subunit of the glutamic
acid decarboxylase enzyme, and morbid obesity was identified
in a French population following a genome-wide scan[48];
however, independent replication in a German population
could not be achieved[49]. Although this example raises
questions concerning the role of GAD2 in obesity, it would
be premature to discount GAD2's involvement entirely[50].
This notion may also apply to additional candidates emerging
from genome-wide scans, as they may also face replication
issues.
Possibly a failure to replicate data is related to the
sample size in which the association was first detected.
Indeed, because the contribution of any given gene to the
phenotype of a complex trait is often minimal, a large
cohort size is required if statistical significance is to be
achieved; however, the caveat is that the more associations
examined, the greater the risk of type-I errors. Statistical
approaches such as linkage of disequilibrium threshold
values and permutation analyses have proven useful, but an
ingenious approach recently described will undoubtedly
provide a template for future association studies[51] .
Using a multi-stage design, in which the number of SNPs
considered is reduced at each step without sacrificing
genome-wide significance, the authors selected the first ten
SNPs for further analysis, and only one, a SNP variant near
the INSIG2 gene, was associated with obesity. While more
common statistical tests (Bonferroni corrections), did not
identify this variant, the multi-stage approach employed by
the authors proved accurate, as this variant was replicated
in four out of five independent populations [44,51].
What can we learn from genetic investigations on obesity
Over the past year important efforts has been invested in
the search for genes that predispose to common obesity,
among these, the major breackthough in obesity research was
the discovery of FTO[52] gene, where variants located in the
first intron of FTO were associated with 1.7 fold increased
risk of obesity, and TCF7 a synembryon-like developmental
gene[52].
It is unclear at the present stage whether counseling and
genetic testing will be integrated into the practice of
obesity prevention and treatment. Before this can occur,
answers to several questions are needed, in addition to a
response to the more general questions such psychological,
social, and ethical. We are able to define the predictive
risk related to obesity for gene variations or mutations in
candidate genes? Although the genetic prediction for
monogenic diseases is very high, the predictive risk is
small and difficult to determine for the common causes of
obesity. We face here more probability than predictive
medicine.
Deficient growth during fetal life and infancy is associated
with an increased risk of obesity, lipids abnormalities,
coronary disease, metabolic syndrome and type 2 diabetes
later in life. This early pattern of growth is associated
with an increased disease risk especially when followed by a
relative gain in body size later in childhood. Genetic
factors are closely involved in growth and disease
pathogenesis and gene-early life environmental interactions
affect health outcomes[53].
Are we able to specifically treat or prevent obesity in
carriers of allelic variants or mutations conferring a
defined risk? Actually, there is no specific and efficient
drug treatment for obesity. Conventional therapy in children
can lead to excessive pressure and inexorably conducts to
worsening obesity. Is it then reasonable to target a
specific population-particularly children-for prevention?
In patients with MC4R mutations, the frequency and relative
risk for obesity (100-fold higher) could lead us to consider
mutation screening at a population scale. MC4R agonists are
now under development and might be used in the future in
patients with decreased melanocortinergic activity. However,
the expression of the disease in mutation carriers is
variable and the penetrance incomplete, emphasizing the role
of environment as well as other genetic contributors.
With all these uncertainties, it seems premature to promote
the detection of the mutation on a large scale. Although
scientists hope that personalized health care based on
genetic profiling will help people recognize their risk and
improve their behavior, additional evidence is needed to
support this possibility. The present revision, as all
narrative review, could be limited for selection search bias.
Neuroendocrinology and genes
Fat mass is an energy storehouse but also an endocrine
organ. A deregulation of the sympathetic nervous system (SNS)
might generate obesity. After stress SNS fibers liberate
neuropeptide Y (NPY) that directly increases visceral fat.
Adrenergic receptors are the major regulators of lipolisis.
In severe obesity the adrenergic receptors subtypes are
differentially expressed in different fat depots. Liver and
visceral fat share a common sympathetic pathway. The
neuroendocrine melanocortinergic system and gastric gherlin
are greatly deregulated in obesity. The specific mutation of
type 4 melanocortin receptor induces early obesity onset,
hyperphagia and insulin resistance. The recently discovered
prohormone convertase mutation simultaneously produces in
1/3 of cases severe gastrointestinal dysfunction and obesity[54,55].
Conclusion
One of the major interests of the of clinical genetic
studies in patients, is to provide breakthroughs in the
understanding of molecular mechanisms involved in body
weight regulation[9]. This fact could provide new tools for
the development of common obesity therapies, an objective
that will be reached in the near future. Predictive genetics
is problematic in common forms of obesity and does not
justify large-scale testing until specific treatments are
not available. However, once these therapies are developed,
genetic testing could allow classification of patients in
subgroups for which the efficacy of different treatments
could be proved[9] .
References
1 Troiano RP, Flegal KM.Overweight children and
adolescents: description,epidemiology, and demographics.
Pediatrics 1998;101:497-504.
2 Flegal KM, Wei R, Ogden C Weight-for-stature compared with
body mass index-for-age growth charts for the United States
from the Centers for Disease Control and Prevention. Am J
Clin Nutr 2002;75: 761-766.
3 Lunshof JE, Pirmohamed M, Gurwitz D. Personalized
medicine: Decades away? Pharmacogenomics. 2006;7:237–241.
4 Hunter DJ. Gene-environment interactions in human diseases.
Nat Rev Genet. 2005;6:287–298.
5 Mutch DM, Wahli W, Williamson G. Nutrigenomics and
nutrigenetics: The emerging faces of nutrition. FASEB J.
2005;19:1602–1616.
6 Kaput J, Ordovas JM, Ferguson L, van Ommen B, Rodriguez RL,
et al. The case for strategic international alliances to
harness nutritional genomics for public and personal health.
Br J Nutr. 2005;94:623–632.
7 Gorman C. Does my diet fit my genes? The new science of
nutrigenomics has some answers. It explains why fat and
caffeine are worse for some than others. Time.
2006;167:69–70.
8 Spiegelman BM, Flier JS. Obesity and the rgulation of
energy balance. Cell 2001;104:531:543.
9 Barsh GS, Farooqi IS, O'Rahilly S. Genetics of body-weight
regulation. Nature 2000;404: 644-651
10 Foufelle F, Ferré P.New perspectives in the regulation of
hepatic glycolytic and lipogenic genes by insulin and
glucose: a role for the transcription factor SREBP-1c.
Biochem J 2002;366: 377-391
11.Ochoa MC, Marti A, Azcona C, Chueca M, Oyarzabal M,
Pelach R, et al. Gene-gene interaction between PPAR gamma 2
and ADR beta 3 increases obesity risk in children and
adolescents. Int J Obes Relat Metab Disord. 2004;28:S37–S41.
12 Argyropoulos G, Rankinen T, Neufeld DR, Rice T, Province
MA, Leon AS, et al. A polymorphism in the human agouti-related
protein is associated with late-onset obesity. J Clin
Endocrinol Metab. 2002;87(9):4198–4202.
13 Martinez JA, Corbalan MS, Sanchez-Villegas A, Forga L,
Marti A, Martinez-Gonzalez
MA. Obesity risk is associated with carbohydrate intake in
women carrying the Gln27Glu beta2-adrenoceptor polymorphism.
J Nutr. 2003;133:2549–2554.
14 Waterland RA, Garza C. Potential mechanisms of metabolic
imprinting that lead to chronic disease. Am J Clin Nutr.
1999;69:179–197.
15 Gillman MW. Epidemiological challenges in studying the
fetal origins of adult chronic disease. Int J Epidem.
2002;31:294–299.
16 Cuestas Montañés E, Achával Geraud A, Garcés Sardiña N,
Larraya Bustos C.Waist circumference, dyslipidemia and
hypertension in prepubertal children. An Pediatr (Barc).
2007;67:44-50
17 Hunter DJ. Gene-environment interactions in human
diseases. Nat Rev Genet. 2005;6:287–298.
18 Mutch DM, Wahli W, Williamson G. Nutrigenomics and
nutrigenetics: The emerging faces of nutrition. FASEB J.
2005;19:1602–1616.
19 Li G, Zhang Y, Wilsey JT, Scarpace PJ. Hypothalamic pro-opiomelanocortin
gene delivery ameliorates obesity and glucose intolerance in
aged rats. Diabetologia. 2005;48:2376–2385.
20 Seeley RJ, Burklow ML, Wilmer KA, Matthews CC, Reizes O,
et al. The effect of the melanocortin agonist, MT-II, on the
defended level of body adiposity. Endocrinology.
2005;146:3732–3738.
21 Molinoff PB, Shadiack AM, Earle D, Diamond LE, Quon CY.
PT-141: A melanocortin agonist for the treatment of sexual
dysfunction. Ann N Y Acad Sci. 2003;994:96–102.
22 Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, et al.
Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium-
3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol-
1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent,
selective, melanocortin subtype-4 receptor agonist. J Med
Chem. 2002;45:4589–4593.
23 Wessells H, Hruby VJ, Hackett J, Han G, Balse-Srinivasan
P, et al. MT-II induces penile erection via brain and spinal
mechanisms. Ann N Y Acad Sci. 2003;994:90–95.
24 Chung WK, Leibel RL. Molecular physiology of syndromic
obesities in humans. Trends Endocrinol Metab.
2005;16:267–272.
25 Goldstone AP. Prader-Willi syndrome: Advances in genetics,
pathophysiology and treatment. Trends Endocrinol Metab.
2004;15:12–20.
26 Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE,
et al. Elevated plasma ghrelin levels in Prader-Willi
syndrome. Nat Med. 2002;8:643–644.
27 Franzese A, Romano A, Spagnuolo MI, Ruju F, Valerio G.
Growth hormone therapy in children with Prader-Willi
syndrome. J Pediatr. 2006;148:846.
28 Carrel AL, Moerchen V, Myers SE, Bekx MT, Whitman BY, et
al. Growth hormone improves mobility and body composition in
infants and toddlers with Prader-Willi syndrome. J Pediatr.
2004;145:744–749.
29 Katsanis N, Lupski JR, Beales PL. Exploring the molecular
basis of Bardet-Biedl syndrome. Hum Mol Genet.
2001;10:2293–2299.
30 Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, et al.
BBS10 encodes a vertebrate-specific chaperonin-like protein
and is a major BBS locus. Nat Genet. 2006;38:521–524.
31 Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson
AL, et al. Comparative genomics and gene expression analysis
identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum
Genet. 2005;77:1021–1033.
32 Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, et al.
Homozygosity mapping with SNP arrays identifies TRIM32, an
E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene
(BBS11). Proc Natl Acad Sci U S A. 2006;103:6287–6292.
33 Eichers ER, Lewis RA, Katsanis N, Lupski JR. Triallelic
inheritance: A bridge between Mendelian and multifactorial
traits. Ann Med. 2004;36:262–272.
34 Rankinen T, Perusse L, Weisnagel SJ, Snyder EE, Chagnon
YC, Bouchard C 2002 The human obesity gene map: the 2001
update. Obes Res 10: 196-243
35 Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V,
Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM,
Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B
1998 A mutation in the human leptin receptor gene causes
obesity and pituitary dysfunction. Nature 392: 398-401
36 Dubern B, Clement K, Pelloux V, Froguel P, Girardet JP,
Guy-Grand B, Tounian P. Mutational analysis of melanocortin-4
receptor, agouti-related protein, and alpha-melanocyte-stimulating
hormone genes in severely obese children. J Pediatr
2001;139: 204-209
37 Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage
disequilibrium in the human genome. Nat Rev Genet.
2002;3:299–309.
38 Slavotinek AM, Searby C, Al-Gazali L, Hennekam RC,
Schrander-Stumpel C, et al. Mutation analysis of the MKKS
gene in McKusick-Kaufman syndrome and selected Bardet-Biedl
syndrome patients. Hum Genet. 2002;110:561–567.
39 Fan Y, Esmail MA, Ansley SJ, Blacque OE, Boroevich K, et
al. Mutations in a member of the Ras superfamily of small
GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet.
2004;36:989–993.
40 Marshall WF, Nonaka S. Cilia: Tuning in to the cell's
antenna. Curr Biol. 2006;16:R604–R614.
41 Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski
RE, et al. Bardet-Biedl syndrome type 4 (BBS4)-null mice
implicate Bbs4 in flagella formation but not global cilia
assembly. Proc Natl Acad Sci U S A. 2004;101:8664–8669.
42 Mak HY, Nelson LS, Basson M, Johnson CD, Ruvkun G.
Polygenic control of Caenorhabditis elegans fat storage. Nat
Genet. 2006;38:363–368.
43 Roche HM, Phillips C, Gibney MJ. The metabolic syndrome:
The crossroads of diet and genetics. Proc Nutr Soc.
2005;64:371–377.
44 Swarbrick MM, Vaisse C. Emerging trends in the search for
genetic variants predisposing to human obesity. Curr Opin
Clin Nutr Metab Care. 2003;6:369–375.
45 Hebebrand J, Friedel S, Schauble N, Geller F, Hinney A.
Perspectives: Molecular genetic research in human obesity.
Obes Rev. 2003;4:139–146.
46 Gallou-Kabani C, Junien C. Nutritional epigenomics of
metabolic syndrome: New perspective against the epidemic.
Diabetes. 2005;54:1899–1906.
47 Mager J, Bartolomei MS. Strategies for dissecting
epigenetic mechanisms in the mouse. Nat Genet.
2005;37:1194–1200.
48 Mutch DM. Identifying regulatory hubs in obesity with
nutrigenomics. Curr Opin Endocrinol Diabetes.
2006;13:431–437.
49 Laird NM, Lange C. Family-based designs in the age of
large-scale gene-association studies. Nat Rev Genet.
2006;7:385–394.
50 Glazier AM, Nadeau JH, Aitman TJ. Finding genes that
underlie complex traits. Science. 2002;298:2345–2349.
51 Slavotinek AM, Searby C, Al-Gazali L, Hennekam RC,
Schrander-Stumpel C, et al. Mutation analysis of the MKKS
gene in McKusick-Kaufman syndrome and selected Bardet-Biedl
syndrome patients. Hum Genet. 2002;110:561–567.
52 Li S, Loss RJ. Progress in the genetics of common obesity:
size matters. Curr Opin Lipidol 2008; 19(2):113-21.
53 Erikson JG. Epidemiology, genes and environment: lessons
learned from the Helsinki birth cohort study. J Intern Med
2007;261:418-25.
54 Garruti G, Cotecchia S, Giampetruzzi F, Giorgino F,
Giorgino R. Neuroendocrine deregulation of food intake,
adipose tissue and gastrointestinal system in obesity and
metabolic syndrome. J Gastrointest Liver Dis
2008;17(2):193-98.
55 Triana Hernández M, Ruiz Álvarez V. Obesity, a wordl
epidemics.Genetic implications. Rev Cubana Invest Biomed
2007;26(2):1-10.

|

