|
Mecanismo de persistencia del
virus rubéola en infección congénita
María P. Adamo
 ,
Mauro S. Pedranti, María J. Calloni Sappag, Marta T. Zapata. ,
Mauro S. Pedranti, María J. Calloni Sappag, Marta T. Zapata.
Revista
Facultad de Ciencias Médicas 2009; 66(Supl.1): 7-14
Instituto de Virología “Dr. J.
M. Vanella”, Facultad de Ciencias Médicas, Universidad
Nacional de Córdoba. Calle Enf. Gordillo Gómez S/N, Ciudad
Universitaria, CP 5016, Córdoba, Argentina.
Introducción
La rubéola es una enfermedad exantemática benigna típica de
la niñez, que generalmente cursa sin presentar
complicaciones y confiere inmunidad protectora de por vida.
La infección en la mujer embarazada en cambio constituye un
grave riesgo para el feto, especialmente durante el primer
trimestre de gestación, por la capacidad teratogénica del
virus. El virus rubéola (RUB) puede inducir un amplio rango
de anomalías congénitas, en conjunto denominadas síndrome de
rubéola congénita (SRC). Mientras la infección postnatal es
limitada por la respuesta inmune del hospedador y se
resuelve en el corto plazo, la rubéola congénita es
persistente. El mecanismo de persistencia viral en la
rubéola congénita no se conoce, pero la infección
persistente se resuelve usualmente entre 1 y 2 años después
del nacimiento (1-2).
Las vacunas a virus atenuado de RUB son efectivas y seguras
y se han utilizado exitosamente en programas de vacunación
por 30 años, lo que convierte a RUB en un atractivo vector
vacunal (3). La dilucidación de las interacciones
virus-célula es importante para comprender y predecir el
comportamiento del virus en su aplicación tecnológica. Por
ello el objetivo de este trabajo es contribuir al
conocimiento de la persistencia de RUB en la infección
congénita, analizando el rol de la apoptosis en la relación
RUB-célula hospedadora.
2 El virus, la rubéola postnatal y la rubéola congénita.
RUB pertenece a la familia Togaviridae, siendo (hasta
ahora) el único togavirus no-arbovirus y único representante
del género Rubivirus. Posee un genoma de ARN infeccioso de
cadena simple protegido por la cápside y una envoltura con
dos glicoproteínas virales. El único hospedador y reservorio
natural de RUB es el hombre y no existe un modelo animal
para la rubéola congénita (4).
La infección por RUB en niños y adultos está caracterizada
por exantema de corta duración, linfoadenopatías, fiebre
moderada y artralgias. Hay una alta proporción de casos
subclínicos (2) y las complicaciones son infrecuentes (5).
RUB se transmite a través de aerosoles respiratorios. La
incubación es seguida por viremia y liberación de partículas
virales infectivas en nasofaringe. La viremia cesa con la
aparición de los anticuerpos circulantes. IgM es la primera
en detectarse y la IgG confiriere protección por el resto de
la vida (1-2; 5-8).
En la primoinfección de una mujer embarazada, los tejidos
placentarios son invadidos durante la viremia materna y la
infección puede transmitirse de la madre al feto (9). La
tasa de infección fetal es cercana a 90% en el primer
trimestre de gestación (5). RUB se disemina ampliamente en
los tejidos fetales (10-11), donde se establece una
infección crónica y no lítica (9).
El 85% de los niños nacidos con rubéola materna confirmada
en el primer trimestre de gestación presenta alguna o varias
de las manifestaciones del SRC (1; 5), que incluyen
patologías transitorias, daños estructurales permanentes y
patologías de emergencia tardía. Las más comunes afectan
sistema cardiovascular, sistema nervioso, ojos y oídos (5;
12).
Los niños con rubéola congénita excretan virus al momento
del nacimiento y una proporción cada vez menor de ellos
continuará haciéndolo los próximos 24 meses (13-15). Los
factores que determinan la duración de la excreción viral no
se conocen.
Los mecanismos teratogénicos tampoco están dilucidado,
aunque resultados directos de la replicación del virus
(crecimiento lento y depolimerización del citoesqueleto)
durante períodos críticos de la ontogenia podrían dar lugar
a las malformaciones del SRC (16-19). También podría
contribuir cierto efecto lítico de RUB en las células
embrionarias / fetales (9; 20) y mediadores de la respuesta
inmune (5; 21). La posible participación del proceso de
muerte celular inducido por RUB en el mecanismo teratogénico
se renovó después al describirse la capacidad de RUB de
inducir apoptosis in vitro (22-23).
La persistencia de RUB en los tejidos fetales indica que ni
los anticuerpos maternos ni los fetales pueden eliminar al
virus in utero, aún cuando ambos pueden neutralizarlo in
vitro (24). Los linfocitos de niños infectados durante la
gestación muestran deficiencia proliferativa in vitro
(25-27), pero producen IgM, IgA e IgG (28-30), por lo que no
está claro cómo escapa RUB a la eliminación y establece una
infección persistente.
3. Hipótesis de persistencia viral en la infección
congénita por virus Rubéola.
Varios mecanismos podrían explicar la persistencia de
RUB en la infección congénita.
3A. Defecto en los mecanismos de defensa del
hospedador.
La inmunotolerancia clásica (con ausencia de anticuerpos) no
es una característica de la rubéola congénita, pero se han
observado diferencias cualitativas en la respuesta de
anticuerpos en las infecciones postnatal y congénita, y
falla en la respuesta proliferativa in vitro de linfocitos
de recién nacidos con rubéola congénita (25-27; 31). Esta
deficiencia indica disfunción de la inmunidad mediada por
células y podría conducir a la persistencia viral por falta
de eliminación de las células infectadas.
3B. La hipótesis clonal.
Considerando la ausencia de potencial citolítico de RUB en
células de cultivos crónicamente infectados que no se podían
“curar” con antisuero específico, y evidencia de que el
virus pasa a las células hijas durante la división celular,
se propuso que se infecta un número limitado de células del
embrión y que las células parentales infectadas dan lugar a
clones de células infectadas, no siendo afectada la
transferencia del virus por los anticuerpos circulantes. Los
clones de células infectadas tendrían un potencial de
duplicación reducido y al ir muriendo resultarían en la
eliminación de la infección congénita (18).
3C. Efectos teratogénicos y persistencia intracelular
del virus independientes.
La disfunción inmune sería consecuencia de la infección
fetal y dependerá del tiempo de gestación al momento de la
infección. La invasión del feto por RUB resulta en una
población de células infectadas que tienen un tiempo de vida
finito. Si la inmunidad celular es deficiente, las células
infectadas sobrevivirán hasta completar su tiempo de vida
determinado. El virus persistirá en tanto las células
infectadas sobrevivan. Así, la duración de la infección está
determinada por el tiempo de vida de la célula infectada y
es influenciada secundariamente por mecanismos inmunológicos
(32).
4. Influencia del background genético de la célula.
Entre 1998 y 2002 se demostró que RUB induce apoptosis
con activación de caspasa 3 en cultivos (Vero, RK13,
neuronas de ratón), que el efecto citopático (ECP) de RUB
está mediado por la apoptosis y que la magnitud de apoptosis
inducida varía entre tipos celulares (21-22; 33-36). Megyeri
y col. (33), observaron apoptosis inducida por RUB en
células Vero y RK13, pero no en dos líneas de fibroblastos
embrionarios humanos (HEL-17 y HEL-18). Muchos virus activan
sensores que desencadenan la apoptosis como respuesta innata
frente a la infección viral (37-38). En este caso la
apoptosis sería un mecanismo de defensa autónomo de la
célula destinado a cortar el ciclo de replicación del virus
y deshacerse de la célula infectada (39-40).
La apoptosis es un proceso clave en el desarrollo
embrionario, en el que algunas células están programadas
para morir, mientras que otras están determinadas para
sobrevivir y proliferar (41-43). Estas células en activa
proliferación tienen sus vías apoptóticas bloqueadas
(44-45), por lo que la expresión normal de oncogenes
promotores de la supervivencia y la proliferación celular
podría impedir la respuesta antiviral autónoma de la célula.
5. Diferencias entre células fetales y de adulto humano y su
respuesta a la infección por virus rubéola.
En cultivos de células Vero, las cepas Gilchrist (prototipo
de laboratorio) y Córdoba (un aislamiento local de 1988 que
carece del historial de pasajes de las cepas atenuadas
prototipos o vacunales) difieren en su capacidad
citopatogénica. A 5 dpi RUB Córdoba induce ~45% de muerte
celular, mientras RUB Gilchrist induce ~30% (36; 46).
Para analizar el rol de la apoptosis en la infección por RUB
se comparó el ECP inducido por RUB Córdoba en células Vero y
células humanas derivadas de embrión y de adulto. Se
establecieron cultivos primarios de fibroblastos fetales
humanos (FFH) proliferantes, explantos de vellosidades
coriónicas (EVC) y cultivos en monocapa de citotrofoblastos
(CTB) de placentas a término, y se utilizaron también
cultivos de células Hs888Lu, fibroblastos diploides
derivados de pulmón humano de individuo adulto (ATCC CCL-211).
RUB induce moderado a marcado ECP (apoptosis) en Hs888Lu,
EVC y en CTB, pero no en FFH (23; 47), y p-Jun [subunidad de
un factor de transcripción implicado en la proliferación
celular (49-51)] no se detectó en células Hs888Lu pero sí en
FFH normales e infectadas (48). Por ello se analizó la
expresión de genes en FFH y Hs888Lu, comparando los niveles
de expresión en estos dos cultivos entre sí y en cultivos
infectados versus controles correspondientes.
El análisis de expresión de genes en cultivos infectados
permitió identificar ~500 genes inducidos o suprimidos por
RUB en FFH y en Hs888Lu (de identidad diferente pero con un
patrón de funciones similar); 5% de ellos participan en la
defensa celular (respuesta innata, citoquinas, quemoquinas),
y la mayoría de los restantes interviene en transducción de
señales, regulación de la trascripción y metabolismo
celular. Del análisis de los genes inducidos y suprimidos
por RUB en células Hs888Lu surge que el patrón de expresión
de genes en estas células promueve la apoptosis luego de la
infección, en la cual la vía del IFN tiene una participación
importante (23). La figura 1 muestra una síntesis
esquemática de estos resultados.
A
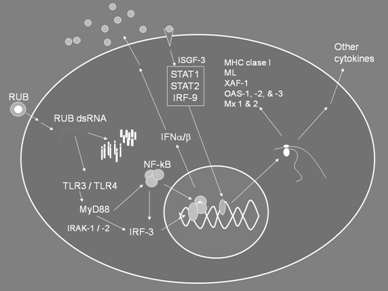
B
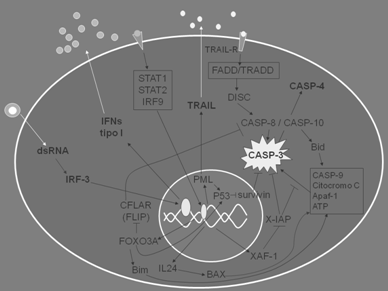
Fig. 1. A. Inducción de IFN en Hs888Lu infectados por RUB.
El ARN viral de doble cadena generado durante la replicación
reconocido por TLR3/4 activa los factores IRF3 y NF-B, que
en el núcleo promueven la transcripción de IFN. El IFN
secretado se une a sus receptores en la superficie celular y
desencadena la respuesta antiviral, incluyendo la expresión
de OAS y MX, las que participan en la degradación de ARN y
proteínas virales. IFN también induce la actividad de
proteínas apoptóticas. B. IFN tipo I induce la transcripción
de genes como FOXO3A, IL24, XAF-1, PML, caspasa (CASP) 10 y
CASP4. CASP10 es una caspasa apical en la cascada enzimática
que caracteriza a la apoptosis; es activada por señales
transducidas desde los llamados “receptores de muerte”
ubicados en la membrana plasmática. En su lado citoplásmico,
estos receptores poseen “dominios de muerte” (DD, por death
domain). Al unir sus ligandos, los receptores reclutan en el
citoplasma, a través de sus dominios DD, moléculas que
también poseen regiones DD, como FADD y TRADD. En la
proximidad de la membrana plasmática se forma entonces del
complejo DISC (death inducing signaling complex), que
transmite la señal de muerte activando CASP8 o CASP10. Estas
activan CASP3, que a su vez activa caspasas efectoras
(CASP4, CASP6, CASP7). La vía intrínseca de la apoptosis
involucra la activación de la CASP3 luego de la disrupción
del potencial de membrana mitocondrial, acompañado de la
liberación hacia el citoplasma de citocromo c. En el
citoplasma, citocromo c, CASP9, Apaf-1 y ATP constituyen el
apoptosoma que activa CASP3. Las vías extrínseca e
intrínseca están conectadas por factores pro-apoptóticos
activados por la vía de los receptores de muere que pueden
desencadenar la salida de citocromo c de la mitocondria, por
ej. Bid.→: activación o inducción; ┤: inactivación o
supresión (52).
En contraste, el patrón general de expresión de genes pro- y
anti-apoptóticos luego de la infección es antagonista de la
apoptosis en las células fetales: genes pro-apoptóticos que
son inducidos por RUB en Hs888Lu no muestran cambios en su
nivel de expresión en FFH o son inducidos en menor magnitud
que en Hs888Lu (BAX, CARD6, CASP10, CASP7, CASP8), mientras
que algunos genes anti-apoptóticos son inducidos (BCL2A1,
BIRC3) y otros pro-apoptóticos son suprimidos (TP53). No
sólo es diferente la respuesta ante la infección por RUB en
los genes que se activan o suprimen en FFH y Hs888Lu, sino
también el patrón de expresión previo a la infección. Las
células fetales tienen sobre-expresados genes anti-apoptóticos
y sub-expresados genes pro-apoptóticos, de tal manera que el
patrón de expresión de genes prevaleciente promueve la
supervivencia en FFH (23). La figura 2 esquematiza
sintéticamente la posible respuesta de células fetales a la
infección por RUB en base a estos resultados.

Fig. 2. La inducción del IFN es parte de la respuesta
antiviral de la célula fetal, sin embargo la fase efectora
de la apoptosis parece estar bloqueada por diversos
mecanismos anti-apoptóticos [AVEN, SURVIVIN, TNFRSF4,
TNFSF18, y TRAIL-R (decoy) están sobre-expresados en FFH y
son anti-apoptóticos; CASP10, Citocromo c y APAF1 son pro-apoptóticos
pero están sub-expresados en FFH con respecto a Hs888Lu]. →:
activación o inducción; ┤: inactivación o supresión (52).
Las células FFH infectadas por RUB muestran además menor
actividad de CASP3 que las células Vero infectadas (53). Más
aún, el tratamiento de cultivos de FFH con varios compuestos
químicos que inducen apoptosis por diferentes vías (actinomicina
D, camptotecina, cicloheximida, dexsametasona y etoposida)
mostró que estas células fetales son relativamente
resistentes a todos ellos excepto cicloheximida (inhibidor
de la síntesis proteica), en contraste con lo que ocurre con
Hs888Lu y Vero (23).
Entre los genes promotores de la supervivencia de la célula
sobre-expresados en FFH destacan integrantes de las vías de
la PI3K (phosphoinositide 3 kinase) y de las MAP-kinasas (MAPK,
mitogen activated protein kinases). En efecto, Akt (quinasa
aguas abajo de PI3K) y ERK (una MAPK) están activas (p-Akt y
p-ERK) en FFH. Si se infectan células FFH con la activación
de Akt y ERK bloqueada por inhibidores específicos, RUB sí
induce apoptosis en ellas. Además, la infección puede
establecerse inicialmente en presencia de inhibidores de la
fosforilación de Akt y ERK, pero luego del segundo día
post-infección el título viral cae significativamente y la
infección no progresa. Similares resultados se obtuvieron
con cultivos FFH persistentemente infectados (53-54; Adamo y
col., manuscrito en preparación). En conjunto, los datos
sugieren que las vías PI3K-Akt y Ras-Raf-MEK-ERK activas en
FFH colaboran en determinar el estado de supervivencia de
FFH y permitirían el establecimiento de la infección
persistente por RUB.
6. Rol de la apoptosis en la infección por virus rubéola.
La capacidad de RUB para inducir apoptosis en distintos
tipos celulares difiere según su grado de diferenciación (y
no proliferante). Además RUB induce la activación de Akt y
ERK en células Vero, BHK-21, Hs888Lu (Adamo y col.,
manuscrito en preparación) y RK-13 (55), aunque sólo de
manera transitoria y previo a la aparición de ECP
(apoptosis) en el cultivo. Ello indica que la apoptosis
inducida por RUB es un mecanismo innato de defensa contra la
infección viral, autónomo de la célula. Como otros virus,
RUB habría encontrado una vía para escapar a este mecanismo
de defensa, prolongando la vida de la célula el tiempo
suficiente para completar su ciclo.
Dado que se observan diferencias en el potencial
apoptogénico de diferentes cepas de RUB, siendo más
citopatogénicas las cepas salvajes que las cepas atenuadas
prototipos (36; 47; 52; Adamo y col., manuscrito en
preparación), el grado de atenuación viral también influye
en su habilidad para sobrepasar el umbral de detección de la
infección e inducción de la respuesta antiviral. Aún no se
sabe qué factores influyen en la capacidad apoptogénica de
RUB. Hasta ahora no se encontraron dominios en las proteínas
virales capaces de interactuar con proteínas celulares para
inducir apoptosis por vía intrínseca, pero experimentos en
los que se bloquea la actividad del IFN sugieren que existe
un mecanismo por el que RUB induce apoptosis independiente
de la vía del IFN (23).
Conclusión
La persistencia de RUB en la infección congénita parece
resultar fundamentalmente de la falla de la respuesta
citotóxica fetal y del tiempo de vida de las células
infectadas, determinado por su patrón de expresión de genes.
Durante la infección embrionaria o fetal, algunas células
infectadas podrían morir por apoptosis, en tanto otras,
determinadas a sobrevivir y proliferar (con sus vías
apoptóticas bloqueadas) no activarían el programa de muerte
como defensa contra virus. En este caso, la expresión normal
de oncogenes promotores de la supervivencia y la
proliferación celular bloquearía la apoptosis inducida por
RUB. Es decir que genes que determinan el destino a
sobrevivir y proliferar de las células durante el desarrollo
embrionario podrían promover la persistencia de RUB en la
infección congénita.
Bibliografía
1. Banatvala J E, Brown D W G. Rubella. Lancet; 2004,
363:1127-1137.[Abstract]
2. Zapata M. Virus de Rubéola. EN Basualdo JA, Cotto CE, De
Torres RA (eds): Microbiología Biomédica. Atlante SRL.
Buenos Aires. 2006. Capítulo 92, p910-917. 3ra ed.
3. Pugachev K V, Tzeng W P, Frey T K. Development of a
Rubella virus vaccine expression vector: use of a
picornavirus internal ribosome entry site increases
stability of expression. J Virol; 2000, 74:10811-10815.
[Full Text]
4. Frey T K. Molecular biology of Rubella virus. Adv Virus
Res; 1994, 44:69-160.
5. Wolinsky J S. Rubella. EN Fields B N, Knipe D M, Howley P
M. et al. (eds): Fields Virology. Lippincott-Raven.
Philadelphia. 1996, p899-929. 3rd ed.
6. Córdoba P A. Mecanismos de neutralización de la
infectividad viral y relación funcional de dos epitopes de
la glicoproteína E1 en distintas cepas de virus Rubéola.
Tesis Doctoral. 1997. Facultad de Ciencias Químicas,
Universidad Nacional de Córdoba.
7. Terry G M, Ho-Terry L, Londesborough P, Res K R.
Localization of the Rubella E1 epitopes. Arch Virol; 1988,
98:189-197.
8. Wolinsky J S, McCarthy M, Allen-Cannady O, Moore W T, Jin
R, Cao S N, Lovett A, Simmons D. Monoclonal antibody defined
epitope map of expressed Rubella virus protein domains. J
Virol; 1991, 65:3986-3994.[Full
text]
9. Tondury G, Smith D W. Fetal rubella pathology. J Pediatr;
1966, 68:867-879.
[Abstract]
10. Alford C A, Neva F A, Weller T H. Virologic and
serologic studies on human products of conception after
maternal rubella. New Engl J Med; 1964, 271:1275-1281.
11. Bellanti J A, Artenstein M S, Olsen L C, Buescher E L,
Luhrs C E, Milstead K L. Congenital rubella:
clinicopathologic, virologic and immunologic studies. Am J
Dis Child; 1965, 110:464-472.
12. Cooper L Z. The history and medical consequences of
rubella. Rev Infect Dis; 1985, 7(Suppl 1):S2-S10.
[Abstract]
13. Katow S. Rubella virus genome diagnosis during pregnancy
and mechanism of congenital rubella. Intervirology; 1998,
4:163-169.
[Abstract]
14. Malathi J, Therese K L, and Madhavan H N. The
association of rubella virus in congenital cataract - a
hospital-based study in India. J Clin Virol; 2001, 23:25-29.
[Abstract]
15. Phillips C A, Melnick J L, You M D, Bayatpour M,
Burkhardt M. Persistence of virus in infants with congenital
rubella and in normal infants with a history of rubella. J
Am Med Ass; 1965, 193:1027-1029.
[Full Text]
16. Rawls W E, Desmyier J, Melnick J L. Virus carrier cells
and virus-free cells in fetal rubella. Proc Soc Exp Biol Med;
1968, 129:479-483.
17. Rawls W E. Viral persistence in congenital rubella. Prog
Med Virol; 1974, 18:273-288.
18. Rawls W E, Melnick J L. Rubella virus carrier cultures
derived from congenitally infected infants. J Exp Med; 1966,
123:795-816.
[Full text]
19. Bowden D S, Pedersen J S, Toh B H, Westaway E G.
Distribution by immunofluorescence of viral products and
actin-containing cytoskeletal filaments in Rubella virus
infected cells. Arch Virol; 1987, 92:211-219.
[Abstract]
20. Neva F A, Alford C A, Weller T H. Emerging perspective
of rubella. Bacteriol Rev; 1964, 28:444-451.
[PubMed Central-Open Access]
21. Adamo M P, Zapata M, Frey TK. Analysis of gene
expression in fetal and adult cells infected with rubella
virus. Virology; 2008, 370:1-11.[PubMed
Central-Open Access]
22. Pugachev K V, Frey T K. 1998. Rubella virus induces
apoptosis in culture cells. Virology 250, 359 – 370.
[Abstract]
23. Duncan R, Muller J, Lee N, Esmaili A, Nakhasi H L.
Rubella virus-induced apoptosis varies among cell lines and
is modulated by Bcl-XL and caspase inhibitors. Virology;
1999, 255:117-128.[Abstract]
24. Cooper L Z, Ziring P R, Ockerse A B, Fedum B A, Kiely B,
Krugman S. Rubella: clinical manifestations and management.
Am J Dis Child; 1969, 118:18-29.
[Abstract]
25. Buimovici-Klein E, Cooper L Z. Cell-mediated immune
response in rubella infections. Rev Infect Dis; 1985, 7(Suppl
1):S123-S128.
[Abstract]
26. Buimovici-Klein E, Lang P B, Ziring P R, Cooper L Z.
Impaired cell-mediated immune response in patients with
congenital rubella: correlation with gestational age at time
of infection. Pediatrics; 1979, 64:620-626.
[Full Text]
27. Fuccillo D A, Steele R W, Hensen S A, Vincent M M, Hardy
J V, Bellanti J A. Impaired cellular immunity to rubella
virus in congenital rubella. Infect Immunity; 1974, 9:81-84.
[Full Text]
28. Enders G. Serologic test combinations for safe detection
of rubella infections. Rev Infect Dis; 1985, 7(Suppl
1):S113-S122.
[Abstract]
29. Grangeot-Keros L, Pillot J, Daffos F, Forestier F.
Prenatal and postnatal production of IgM and IgA antibodies
to Rubella virus studied by antibody capture immunoassay. J
Infect Dis; 1988, 158:138-143.
[Abstract]
30. Ueda K, Tokugawa K, Fukushige J, Yoshikawa H, Nonaka S.
Hemagglutination inhibition antibodies in congenital rubella
syndrome: a 17-years follow-up in the Ryukyu Islands. Am J
Dis Child; 1987, 141:211-212.
[Abstract]
31. Olson G B, Dent P B, Rawls W E, South M A, Montgomery J
R, Melnick J L, Good R A. Abnormalities of in vitro
lymphocyte responses during rubella virus infections. J Exp
Med; 1968, 128:47-68.
[PubMed central-Open Access]
32. Simons M J. Congenital rubella: an immunological paradox?
Lancet; 1968, 2:1275-1278.
33. Megyeri K, Berencsi K, Halazonetis T D, Prendergast G C,
Gri G, Plotkin S A, Rovera G, Gonczol E. Involvement of a
p53-dependent pathway in rubella virus-induced apoptosis.
Virology; 1999, 259:74-84.
[Abstract]
34. Hofmann J, Pletz M W, Liebert U G. Rubella virus-induced
cytopathic effect in vitro is caused by apoptosis. J Gen
Virol; 1999, 80:1657-1664.
[Full Text]
35. Domegan L M, Atkins G J. Apoptosis induction by the
Therien and vaccine RA27/3 strains of rubella virus causes
depletion of oligodendrocytes from rat neural cell cultures.
J Gen Virol; 2002, 83:2135-2143.
[Full Text]
36. Martinez L D, Zapata M T. Apoptosis induction by the
infection with Gilchrist strain of rubella virus. J Clin
Virol; 2002, 25:309-315.[Abstract]
37. O’Brien V. Viruses and apoptosis. J Gen Virol; 1998,
79:1833-1845.
[Full Text]
38. Roulston A, Marcellus R, Branton P E. Viruses and
apoptosis. Annu Rev Microbiol; 1999, 53:577-628.
[Abstract]
39. Everett H, McFadden G. Apoptosis: an innate immune
response to virus infection. Trends Microbiol; 1999,
7:160-165.
[Abstract]
40. Koyama A H, Adachi A. Physiological significance of
apoptosis during animal virus infection. Int Rev Immunol;
2003, 22:341-359.[Abstract]
41. Ellis R E, Yuan J, Horvitz H R. Mechanisms and functions
of cell death. Ann Rev Cell Biol; 1991, 7:663-698.
42. Jurisicova A, Acton B M. Deadly decisions: the role of
genes regulating programmed cell death in human
preimplantation embryo development. Reproduction; 2004,
128:281-291.
43. Oligny L L. Human molecular embryogenesis: an overview.
Pediatr Dev Pathol; 2001, 4:324-343.
[Abstract]
44. Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein
U, Tian J, Karin M, Angel P, Wagner E F. Control of cell
cycle progression by c-jun is p53 dependent. Genes Dev;
1999, 13:607-619.[PubMed
Central Open-Access]
45. Shaulian E, Schreiber M, Piu F, Beeche M, Wagner E,
Karin M. The mammalian UV response: c-jun induction is
required for exit from p53-imposed growth arrest. Cell;
2000, 103:897-907.
[Abstract]
46. Adamo M P, Leon Monzon M, Cuffini C, Pedranti M, Zapata
M. Detection of rubella-virus-induced apoptosis in Vero cell
cultures with hematoxylin and eosin staining. Rev Argent
Microbiol; 2002, 34:177-185.
[Abstract]
47. Adamo P, Asis L, Silveyra P, Cuffini C, Pedranti M,
Zapata M. Rubella virus does not induce apoptosis in primary
human embryo fibroblast cultures: a possible way of viral
persistence in congenital infection. Viral Immunol;
2004,17:87-100.
[Abstract]
48. Adamo M P, Zapata M, Frey T. Análisis de expresión de
genes en células de embrión y de adulto humano infectadas
por el virus Rubéola: implicancias para la persistencia
viral en la infección congénita. XIII Jornadas de Jóvenes
Investigadores de la Asociación de Universidades del Grupo
Montevideo. Argentina, 31 Ago.-2 Sept. 2005. Trabajo
completo en CD. Resúmenes ND1007, p142.
49. Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath
I, Guertl B, Zenz R, Wagner E F, Zatloukal K. Functions of
c-jun in liver and heart development. J Cell Biol; 1999,
145:1049-1061.
50. Sabapathy K, Hochedlinger K, Nam S J, Bauer A, Karin M,
Wagner E F. Distinct roles for JNK1 and JNK2 in regulating
JNK activity and c-Jun dependent cell proliferation. Mol
Cell; 2004, 15:713-725.[Abstract]
51. Sabapathy K, Wagner E F. JNK2 a negative regulation of
cellular proliferation. Cell Cycle; 2004, 3:1520-1523.[Full
Text]
52. Adamo M P. Estudio de un mecanismo de persistencia del
irus rubéola en la rubéola congénita. Tesis Doctoral. 2006.
Facultad de Ciencias Médicas, Universidad Nacional de
Córdoba.
53. Adamo M P, Pedranti M, Zapata M, Frey T. Participación
de vías celulares de supervivencia y proliferación en la
infección por virus Rubéola. XXVII Reunión Científica Anual
de la Sociedad Argentina de Virología. Córdoba, Argentina,
2-5 Dic. 2007. Libro de Actas p19, resumen 22462.
54. Adamo M P, Pedranti M, Zapata M. Las vías PI3K-Akt y
Ras-Raf-MEK-ERK bloquean la apoptosis inducida por el virus
Rubéola en células fetales humanas: un modelo de
persistencia viral en la infección congénita. XIV Jornadas
de Jóvenes Investigadores de la Asociación de Universidades
del Grupo Montevideo. Brasil, 13-15 Sept. 2006. Trabajo
completo en CD. Libro de Actas p502, resumen ND7010.
55. Cooray S, Jin L, Best J M. The involvement of survival
signalling pathways in Rubella-virus induced apoptosis.
Virol J; 2005, 2:1.
[Full Text]

|

