|
REVISIÓN
Manifestaciones
clínicas del síndrome antifosfolípido
Paula Alba, Pablo Astesana, Alejandra Babini.
Marcelo Yorio
Revistad de la Facultad de Ciencias Médicas
2010, 67(1): 40-50
* Postgrado de
Reumatología del Hospital Córdoba. Cátedra de Medicina I.
UHMI 3. ** Servicio Reumatología Hospital Italiano.
Autor correspondiente:
paulaalba@yahoo.com
INTRODUCCIÓN
En el comienzo de la década de los 80, el Dr. Graham Hughes
describió un síndrome clínico caracterizado por trombosis,
abortos recurrentes, enfermedad neurológica y la presencia
de los anticuerpos antifosfolípidos (AAF)
(1).
En 1983, era difícil predecir el interés que la
determinación de los anticuerpos anticardiolipinas (ACA) iba
a generar en los años siguientes
(2). Es frecuente la
descripción de nuevos autoanticuerpos en el Lupus
eritematoso sistémico (LES) y podía parecer que los ACA no
iban a merecer sino un interés pasajero. Sin embargo, su
asociación con una amplia combinación de síntomas que
incluyen fundamentalmente trombosis venosas y arteriales,
pérdidas fetales y trombocitopenia, entre otros han atraído
hacia ellos la mirada de investigadores de los más diversos
ámbitos de la medicina
(1,2,3).
El término síndrome antifosfolípido (SAF) ó Síndrome de
Hughes se aplica a un estado de hipercoagulabilidad que
predispone a trombosis venosa, arterial o ambas asociado a
la presencia de AAF como los ACA y el anticoagulante
lúpico(AL). Este síndrome puede presentarse en forma
aislada, SAF, o en asociación con enfermedades del tejido
conectivo, principalmente LES.
En el consenso Internacional en Sapporo (Japón) se
publicaron los criterios preliminares de clasificación del
SAF
(Cuadro 1) (4).
|
 |
CUADRO 1. CRITERIOS PRELIMINARES DE
CLASIFICACIÓN DEL SAF (SAPPORO 1998) |
Recientemente, estos criterios fueron actualizados
agregándose al criterio de laboratorio los anticuerpos antiß
2 glicoproteína I (ABGPI) confirmados en 2 ocasiones
separados de 12 semanas
(Cuadro 2)
(5).
|
 |
CUADRO 2.
CRITERIOS REVISADOS DE CLASIFICACIÓN DE SAF SYDNEY
(2004) |
Sin embargo, numerosos hallazgos clínicos del SAF no están
incluidos en estos criterios y esto no debe hacer que el
diagnóstico no sea considerado si otras causas han sido
excluidas y existe una alta sospecha clínica.
Si bien la trombosis y la pérdida fetal recurrente así como
la trombocitopenia moderada son uno de los principales
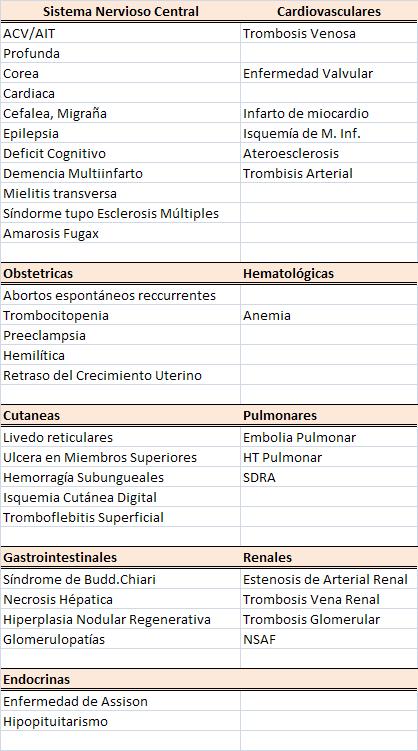
hallazgos, la lista de manifestaciones clínicas es enorme y
numerosos sistemas pueden estar afectados
(Cuadro 3).
|
 |
CUADRO 3.
MANIFESTACIONES CLÍNICAS DEL SAF |
AAF
ACA
Los ACA son positivos en alrededor del 80% de los pacientes
con SAF, mientras que el AL en el 20%, y ambos en
aproximadamente el 60 % de los casos
(6). Sin embargo, ambas
pruebas deben realizarse en los pacientes con sospecha de
SAF.
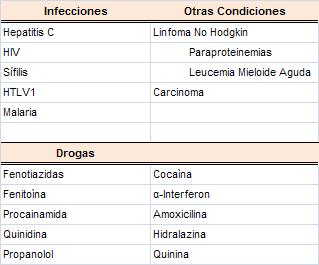
Los ACA pueden ser positivos en una gran variedad de
condiciones que incluyen enfermedades del tejido conectivo,
drogas, neoplasias e infecciones
(Cuadro 4) (7, 8,9).
|
 |
CUADRO 4.
CONDICIONES ASOCIADAS CON AAF |
Sin embargo en éstas últimas, el isotipo predominante es
habitualmente Ig M, en bajos títulos y generalmente no
asociada a trombosis.
La presencia de ACA ha sido descripta en un 44% y la de AL
en un 33% en un estudio de 1000 pacientes con LES, mientras
que sólo el 25 al 35% de pacientes desarrollaron SAF y su
expresión clínica fue independiente de la actividad del LES
(10).
Los ACA son uno de los principales factores de riesgo para
el primer episodio de trombosis venosa profunda y trombosis
venosa recurrente
(11,12).
Finazzi et al. realizaron un estudio prospectivo de 360
pacientes no seleccionados con AL positivo, con y sin ACA
positivos. Ellos demostraron que los ACA > a 40 GPL en
pacientes con eventos trombóticos previos eran factores
predoctores independientes de un nuevo evento vascular
trombótico
(13). Por otra parte,
Escalante et al. y Levine y col. encontraron que no sólo la
presencia sino también los títulos de los ACA son
importantes para identificar un grupo de mayor riesgo de
eventos recurrentes
(14,15).
Las complicaciones obstétricas junto con la trombosis son
una de las principales manifestaciones del SAF. Un
meta-análisis reciente analizó la relación entre los
diferentes los diferentes AAF con la pérdidas del embarazo
en mujeres sin enfermedad autoinmune conocida. La presencia
de AL tuvo una fuerte asociación con la pérdida fetal
recurrente, mientras que los ACA Ig G e Ig M fueron
claramente asociados con las pérdidas recurrentes antes de
la semana 24, y los ACA Ig G con los abortos tempranos. Por
el contrario, los ABGPI no mostraron asociación con ninguna
forma de pérdida del embarazo recurrente
(16).
AL
El AL es identificado por las la prolongación de los tiempos
de coagulación. Un número de pruebas necesitan ser
demostradas: la prolongación del tiempo de coagulación
fosfolípidos dependiente, la evidencia de la inhibición
demostradas por las pruebas de corrección, la evidencia de
dependencia de fosfolípido y la exclusión de la inhibición
específica de algún factor de la coagulación. La presencia
de AL es más específica que los ACA para el SAF, aunque
menos sensible. En este aspecto, un metanálisis que evaluó
el riesgo de trombosis venosa en pacientes con LES, demostró
que los pacientes con AL positivo tenían un riesgo seis
veces mayor de tener un evento trombótico que los negativos
(17).
Posteriormente, estos datos fueron confirmados por otro
metaanálisis de AAF y trombosis venosa en pacientes sin
enfermedad autoinmune concluyendo que el AL es un predictor
más específico de trombosis que los ACA
(18).
OTROS AAF
La mayoría de los anticuerpos asociados al SAF y detectados
en pruebas de ACA y AL están dirigidos contra la β2-GPI o la
protrombina. Estudios recientes indican que estos
anticuerpos pueden también ser detectados utilizando
antígenos proteicos purificados en ausencia de fosfolípidos(19,20).Los
estudios clínicos iniciales de los ELISA de ABGPI sugieren
que la positividad de estas pruebas éstos esta estrechamente
asociada a las manifestaciones clínicas del SAF que los
ELISA de ACA convencional
(21).Por otra parte, los
ABGPI también identificarían un pequeño número de pacientes
que tienen manifestaciones clínicas de SAF pero que son
negativos a las pruebas convencionales
(22).
En los nuevos criterios clínicos y de laboratorio los ABGPI
Ig. G e Ig M fueron incluidos por consenso
(5).
Sin embargo, debido a la falta de estandarización, su
aplicación de rutina como herramientas diagnósticas para el
SAF permanece todavía cuestionada.
La protrombina es un blanco antigénico común de los AAF. Los
anticuerpos anti-protrombina pueden ser detectados por
diferentes métodos y una reciente revisión no demostró una
asociación con trombosis
(23).
Sin embargo, Bertolaccini et al. demostraron que éstos
fueron positivos en algunos pacientes con manifestaciones de
SAF que fueron negativos para AL, ACA y ABGPI , sugiriendo
que éstos podrían ser útiles en pacientes que son negativos
a las pruebas de rutina
(24,25).
Numerosos autoanticuerpos han sido descriptos en pacientes
con SAF, incluyendo anti anexina V, precalicreína, factor XI,
heparina, factor XII, y trombina. Sin embargo, la asociación
con el SAF, su significancia clínica, así como la
estandarización de los mismos, están lejos de ser
establecidas.
MANIFESTACIONES CLINICAS
Los AAF se asocian con trombosis que pueden ocurrir a
cualquier nivel del territorio vascular arterial y venoso y
pueden afectar a vasos de todos los tamaños. A nivel venoso,
las trombosis más frecuentes son en las de las extremidades
inferiores y se acompañan a menudo de embolismo pulmonar. Se
estima que alrededor de un 19% de los pacientes que
presentan trombosis venosas profundas y/o tromboembolismo
pulmonar tienen un SAF con positividad para el AL y/o ACA.
El embolismo pulmonar puede originarse de vena cava
inferior, venas renales, vegetaciones de la válvula
tricuspidea o trombosis intracardíaca derecha
(38, 39,40). El embolismo
pulmonar recurrente puede conducir a hipertensión pulmonar
secundaria y es la principal causa de hipertensión pulmonar
en el SAF
(41). La prevalencia de
hipertensión pulmonar ha sido estimada en el 1,8% de
pacientes con SAF asociado a enfermedades autoinmunes y en
el 3,5% con SAF
(42).
También se han descrito trombosis venosas a nivel de venas
axilares, oculares, renales, hepáticas y de la cava
inferior.
La trombosis de los senos venosos cerebrales es una
manifestación menos frecuente y mas común en mujeres
particularmente entre los 20 y 35 años de edad, y aparece
durante el embarazo, puerperio y durante la ingesta de
anticonceptivos orales
(43).
Christopher y col estudiaron 31 pacientes con TVSC
detectando AAF en un 22.6% comparado con el 3,2% de los
controles, sugiriendo por lo tanto que los AAF son un
marcador de riesgo para esta entidad
(44).
El SAF, en la actualidad, es una de las causas más
frecuentes de síndrome de Budd-Chiari y la manifestación
hepática más frecuente
(45).
Recientemente Jung Hoon y col evaluaron los hallazgos
tomográficos de las manifestaciones abdominales en el SAF.
De los 14 pacientes con SAF que fueron estudiados con
tomografía abdominal, el 72% presento compromiso del sistema
venoso, el 14% compromiso del sistema arterial y el 14% de
ambos. La vena cava inferior y la vena hepática fueron los
vasos mas frecuentemente comprometidos. El SAF debería ser
incluido como una de las posibilidades diagnósticas en
pacientes con síndrome de Budd-Chiari o cuando los hallazgos
tomográficos demuestran infartos en múltiples órganos
(46).
Otras manifestaciones hepáticas como trombosis de las venas
hepáticas, infartos intrahepáticos e hiperplasia nodular
regenerativa
(47,48).
Los AAF se han asociado también a la producción de trombosis
de venas adrenales que pueden conducir a una enfermedad de
Addison
(49). Arnason et al
publicaron 27 casos de insuficiencia suprarenal asociada a
la presencia de AAF. Por otra parte, algunos casos de
hipopituitarismo se han asociado a SAF
(50,51). Uno de estos
pacientes presentaba en la RNM la imagen de “silla turca
vacía” que representa el hallazgo neuroradiológico de
necrosis pituitaria
(52).
Las trombosis arteriales constituyen otra de las
manifestaciones más importantes del SAF. La isquemia
cerebral asociada con la presencia de AAF es la
manifestación trombótica arterial más frecuente
(53,54).
La edad de presentación de la isquemia cerebral asociada a
AAF es mas temprana comparada con la población que
habitualmente la presenta
(55)
y puede afectar cualquier territorio vascular
(56).
Las imágenes de la resonancia magnética muestran diversas
alteraciones que van desde una sola lesión isquémica a
infartos múltiples diseminados
(57,58).
En algunos pacientes, las trombosis cerebrales recurrentes
provocan un cuadro de demencia multiinfarto con síntomas
psiquiátricos como forma de presentación.
Las lesiones cardíacas más frecuentemente asociadas a los
AAF son las trombosis de las arterias coronarias, que
conducen a la aparición de angina e infarto de miocardio, y
las vegetaciones en las válvulas cardiacas, que pueden
provocar disfunciones valvulares, embolias cerebrales e,
incluso, cuadros sugestivos de endocarditis infecciosa
(59,60).
La afectación valvular cardíaca es frecuente en pacientes
con SAF
(59,61-63). Un
metaanálisis de estos estudios ha mostrado que el 48% de los
pacientes con AAF positivos y LES tenían compromiso valvular
comparado con el 21% sin AAF
(60). En la mayoría de
los pacientes con AAF el compromiso valvular tiene poco
significado hemodinámico o no causa enfermedad manifiesta.
La válvula mitral es la más afectada, seguida por la aórtica
y el engrosamiento valvular es la lesión más frecuente
detectada por ecocardiografía en pacientes con SAF. La
consecuencia hemodinámica más importante es la insuficiencia
mitral que ocurre en el 22% de los pacientes con SAF y en el
26% de los pacientes con SAF asociado a LES. La
insuficiencia aórtica ocurre en el 6 y el 10% de los
pacientes respectivamente. Las lesiones de estenosis tanto
mitral como aórtica son poco comunes y cuando aparecen van
acompanadas de insuficiencia de las mismas
(60).
Recientemente el Comité de expertos reunidos en Sydney
propuso definiciones de enfermedad valvular cardíaca
asociada a SAF, aunque ésta no fue incluida como criterio
(5).
La patogenia del compromiso valvular es desconocida. Se ha
postulado que los AAF pueden causar un daño valvular o
endotelial directo. Amital et al. describieron el depósito
de ACA en la superficie endotelial de la válvula
(64)
y Garcia Torres et al. postularon la hipótesis de la
interacción entre AAF con factores locales proponiendo que
estos pudieran conducir al daño endocárdico resultando en
trombosis superficial, infiltración mononuclear y fibrosis
(65).
También han sido descritos casos de miocardiopatía aguda o
crónica por trombosis en la microcirculación miocárdica,
trombos intracavitarios así como anormalidades
ecocardiográficas en la relajación y en la fase del llenado
rápido sin evidencia clínica de enfermedad cardíaca
(66-70).
Las trombosis arteriales también pueden afectar a las
arterias retinianas, mesentéricas y arterias periféricas.
Otro síntoma que puede asociarse a la presencia de AAF es la
hipertensión arterial. (HTA). Su presencia es un signo común
asociado a compromiso al compromiso del tronco así como
microvascular de los vasos renales, y clínicamente ésta
puede ir desde formas leves hasta HTA maligna. Nochy et al.
analizaron 16 pacientes con SAF y compromiso renal
encontrando que la HTA estaba presente en el 94% de los
pacientes. Esta fue severa en el 31% de los pacientes y
maligna en el 12%. 14 pacientes tenían insuficiencia renal,
la cual fue leve en la mayoría de los casos. Todos los
pacientes mostraron lesiones vasculares significativas en la
biopsia renal con lesiones oclusivas de los pequeños vasos
(71).La HTA puede ser
también secundaria a compromiso de grandes vasos y la
estenosis de la arteria renal ha sido descripta en pacientes
con SAF. En una de las primeras descripciones Ostuni et al.
reportaron una niña de 13 años con LES e HTA severa y
estenosis renal bilateral
(72).
Recientemente Sangle et al. describieron la presencia de
estenosis de la arteria renal en un 26% de 91 pacientes con
SAF en pacientes con HTA no controlada y lábil. Estos
autores evaluaron además el efecto del tratamiento
anticoagulante, no encontrando reestenosis y permaneciendo
con función renal estable y buen control de su tensión
arterial con un RIN > 3. Todos estos reportes confirman que
el SAF puede ser una causa significativa de estenosis de la
arteria renal e HTA severa
(73,74).
Daugas et al. observaron las lesiones antes mencionadas
definidas hoy como NSAF en pacientes con LES, especialmente
con SAF asociado, y éstas eran independientes de la
nefritis.
Un aspecto de gran importancia es la aparición en algunos
pacientes con AAF, de un cuadro trombótico de tipo
multisistémico y de curso muy grave denominado actualmente
síndrome antifosfolípido catastrófico. Asherson y col
identificaron los primeros pacientes con SAF catastrófico
publicados en la literatura. El 66% eran mujeres, con un
promedio de 36 años y el 55% de ellos desarrolló un
compromiso multiorgánico agudo secundario a trombosis
múltiples. Algunos factores precipitantes podrían ser las
infecciones, drogas, procedimientos quirúrgicos o abandono
de la anticoagulación. La presentación clínica a menudo
compromete a múltiples órganos en un corto período de
tiempo. Este desorden se caracteriza por una
microvasculopatía trombótica difusa que afecta con
predilección al riñón, pulmón, cerebro, corazón, piel y
tracto gastrointestinal. El tratamiento es empírico
incluyendo anticoagulación, inmunosupresión y plasmaféresis;
la mortalidad ocurre en aproximadamente el 50% de los
pacientes
(75,76).
La trombocitopenia es relativamente frecuente en pacientes
con AAF. Es de curso crónico, leve y rara vez se asocia a
complicaciones hemorrágicas
(77,78).
La trombosis venosa o arterial puede presentarse en
presencia de trombocitopenia, sin embargo dos estudios
retrospectivos han revelado menor frecuencia de eventos
trombóticos cuando las plaquetas son inferiores a 50.000(79,80).
Cuadrado et al. evaluaron retrospectivamente la presencia de
trombocitopenia en 171 pacientes con SAF, y el 23,4%
presentó trombocitopenia que no se asoció con ninguna otra
manifestación clínica ni de laboratorio característica
(81).
El papel patogénico de los AAF en la trombocitopenia es
todavía poco claro. Recientes datos han mostrado la
presencia de anticuerpos antiplaquetarios en pacientes con
SAF. Galli y col encontraron que el 40% de pacientes con SAF
tienen anticuerpos antiGPIIB-IIIA o GOIb/IX o ambos. La
presencia de estos anticuerpos tuvo una correlación
significativa con la trombocitopenia
(82).
Recientemente el comité de expertos reunidos en Sydney
propuso una definición de trombocitopenia asociada al SAF,
no siendo incluida como criterios de clasificación
(5). Algunos pacientes
con AAF y trombocitopenia también desarrollan anemia
hemolítica con test de Coombs directo positivo. Esta
asociación ha sido clásicamente denominada síndrome de
Evans. La presencia de test de Coombs positivo ha sido
observada tanto en SAF primario como secundario. En el SAF
primario, el test de Coombs fue positivo en el 14% y la
anemia hemolítica estuvo presente en un 4% de 70 pacientes
estudiados
(82).
Los mecanismos patogénicos de la anemia hemolítica
relacionada con AAF no están claros. Los AAF podrían unirse
directamente a la membrana de los eritrocitos, pero el
antígeno expresado en la superficie de estas células es aún
desconocido. La necrosis de la médula ósea es un síndrome
que conduce a una pancitopenia severa. Ha sido descrito en
numerosas condiciones y recientemente se ha asociado a la
presencia de AAF
(83,84).
La epilepsia y la corea son manifestaciones menos frecuentes
del SAF
(85, 86,87).
La crisis epiléptica se asociaron con la presencia de ACA
tipo IgG en pacientes con LES
(88). Cervera et al.
analizaron 50 pacientes con corea y encontraron que el 58%
de los pacientes tenían diagnóstico de LES y un 30% de SAF.
La mayoría de los pacientes presentaron un solo episodio de
corea que fue bilateral en el 55% y que mostraron infartos
cerebrales en la RNM en el 35% (87). La mielopatía
transversa, aunque rara, está estrechamente relacionada con
la presencia de AAF
(89, 90, 91,92). Los
síndromes clínicos que simulan esclerosis múltiple han sido
también descritos en el SAF
(91,93).
Cuadrado et al. Reportaron 27 pacientes cuyo diagnóstico
inicial fue esclerosis múltiple y que fueron derivados por
síntomas que sugerían una enfermedad del tejido conectivo o
hallazgos atípicos de esclerosis múltiple. No encontraron
diferencias ni en el examen clínico ni en el laboratorio
entre ambas entidades y cuando comparamos las RNM
cerebrales, estas mostraban una puntuación de mayor
severidad en pacientes con esclerosis múltiple, pero
consideradas individualmente no fue posible distinguir entre
ambos diagnósticos. Una historia clínica con antecedentes de
trombosis, pérdida fetal o la localización poco
característica de las lesiones así como la respuesta a la
anticoagulación constituyen algunos puntos que pueden ayudar
al diagnóstico diferencial
(93).
La migraña se observa frecuentemente en pacientes con SAF y
suele preceder a este último diagnóstico en años. Sin
embargo, estudios prospectivos no han demostrado una
asociación estadísticamente significativa entre migraña y la
presencia de AAF
(91, 94,95).
La lìvedo reticularis (LR) es uno de los signos físicos que
con más frecuencia se asocia a la presencia de AAF. La LR se
caracteriza por un colorido violáceo oscuro, con patrón
reticular que puede aparecer tanto en miembros superiores
como inferiores. Esta puede ser transitoria y fisiológica,
que aparecería con la exposición al frío y con un patrón
regular y simétrico. El patrón de LR asociado al SAF es
generalmente asimétrico, diseminado, irregular con amplias
ramificaciones, aunque algunos pacientes presentan un patrón
más regular fino y simétrico .En un estudio prospectivo de
pacientes con LR el 43% tenían ACA
(96).
Algunas observaciones sugieren que un subgrupo de pacientes
con síndrome de Sneddon (LR mas afectación neurológica)
pueden tener SAF (97,
98,99).
Las úlceras cutáneas son otro de los hallazgos más
frecuentes. El 30% de los pacientes del estudio de la
Clínica Mayo presentaron úlceras cutáneas dolorosas, de
bordes irregulares localizados en el área pretibial y del
tobillo
(100).
Una gran variedad de lesiones que incluyen máculas
eritematosas, nódulos dolorosos, hemorragias subungueales y
anetoderma han sido descritos asociados al SAF
(101-104).
Las manifestaciones obstétricas del SAF incluyen abortos
recurrentes, pérdidas fetales, retraso en el crecimiento
intrauterino, prematuridad, desprendimiento placentario y
preeclampsia. El consenso Internacional de Sapporo y Sydney
han definido los criterios de clasificación obstétricos
(Cuadro 1 y 2) (4,5).
La pérdida fetal es el hallazgo obstétrico más
característico. Estas pérdidas pueden ocurrir en cualquier
momento del embarazo pero alrededor del 50% de las pacientes
con SAF experimentan pérdida fetal en el segundo y tercer
trimestre del embarazo
(105). En mujeres con
historia de gestaciones normales solo un 2% tienen o AL
positivo o ACA y menos de un 0.2% tienen títulos altos de
estos anticuerpos
(106,107).
Se considera, por tanto, que la determinación de estos
anticuerpos en mujeres embarazadas sin antecedentes
obstétricos patológicos no este indicada. La historia de los
embarazos previos es importante para determinar la
significación de una prueba positiva para anticuerpos
antifosfolípido. Se estima que si una paciente con lupus
tiene AL o al menos títulos medios del isotipo IgG de ACA,
el riesgo de aborto espontáneo en el primer embarazo es de
un 30% y si tiene una historia de al menos dos pérdidas
fetales previas el riesgo aumenta hasta el 70% en el
siguiente embarazo
(108-111).
En conclusión, el SAF puede afectar cualquier órgano y
sistema, y el medico internista debería incluir esta entidad
dentro de los diagnósticos diferenciales.
BIBLIOGRAFIA
1. Hughes GRV. Trombosis, abortion, cerebral disease and
lupus anticoagulant. Br Med J 1983; 287:1088-1089.[Full
text]
2. Harris EN, Gharavi AE, Boey ML et al. . Anticardiolipin
antibodies: deteccion by radioimmunoassay and association
with thrombosis in systemic lupus erythematosus. Lancet
1983; ii:1211-1214.[Full
Text]
3. Khamashta MA , Asherson RA. Hughes Syndrome-Antiphospholipid
antibodies move closer to thrombosis in 1994. Br J Rheumatol
1995; 34:493-494.
[Full
Text]
4. Wilson WA, Gharavi AE, Koike T et al. International
Consensus Statement on preliminary classification criteria
for definitive Antiphospholipid Síndrome.Report of an
international workshop. Arthritis Rheum 1999;42:1309-1311.
5. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL,
Cervera R et al. International Consensus statement on an
update of the clasification criteria for definitive
antiphospholipid syndrome. J Thromb Haemost 2006; 4 :
295-306.
[Full Text]
6. Cervera R, Piette JC, Font J et al. Antiphospholipid
syndrome: Clinical and immunological manifestations and
patterns of disease expression in a cohort of 1000 patients.
Arthritis Rheum 2002;46: 1019-1027.
[Full Text]
7. Harris EN, Gharavi AE, Wasley GD, Hughes GR. Use of an
enzyme-linked immunosorbent assay and of inhibition studies
to distinguish between antibodies to cardiolipin from
patients with syphilis or autoimmune disorders. J Infect Dis
1988; 157:23-31.
8. Galvez J, Martin I, Merino D, Pujol E. Thromboplebitis in
a patient with acute Q fever and anticardiolipin antibodies.
Med Clin (Barc)1997;108: 396-7.
9. Intrator L, Oksenhendler E, Desforges L, Bierling P.
Anticardiolipin antibodies in HIV infected patients with or
without thrombocytopenic purpura. Br J Haematol 1988;
68:269-70.
[Abstract]
10. Love PE, Santoro SA : Antiphospholipid antibodies:
Anticardiolipin and the lupus anticoagulant in SLE and in
non-SLE disorders. Ann Intern Med 1990; 112: 682-698.
[Abstract]
11. Ginsburg , Liang MH, Newcomer L et al. Anticardiolipin
antibodies and the risk for ischemic stroke and venous
thrombosis. Ann Intern Med 1992;117:997-1002.
[Abstract]
12. Schulman S, Svenungsson E, Granqvist S. Anticardiolipin
antibodies predict early recurrence of thromboembolism and
death amomg patients with venous thromboembolism following
anticoagulant therapy. Duration of Anticoagulation Study
Group. Am J Med 1998;104:332-338.
[Abstract]
13. Finazzi G, Brancaccio V, Moia M et al. Natural history
and risk factors for thrombosis in 360 patients with
antiphospholipid antibodies : a four-year prospective study
from the Italian Registry. Am J Med 1996;100:530-36.[Abstract]
14. Escalante A, Brey RL, Mitchell BD Jr, Dreiner U.
Accuracy of anticardiolipin antibodies in identifying a
history of thrombosis among patients with systemic lupus
erythematosus. Am J Med 1995; 98: 559-565.[Abstract]
15. Levine SR, Salowich-Palm L, Sawaya KL, et al. Ig G
anticardiolipin antibody titer >40 GPL and the risk of
subsequent thrombo-occlusive events and death. A prospective
cohort study. Stroke 1997;28: 1660-65.[Abstract]
16. Opatrny L, David M, Kahn SR, et al. Association between
antiphospholipid antibodies and recurrent fetal loss in
women without autoimmune disease: a metaanalysis. J
Rheumatol 2006; 33: 2214-21.
[Abstract]
17. Wahl DG, Guillemin F, de Maistre E et al. Risk for
venous thrombosis related to antiphospholipid antibodies in
systemic lupus erythematosus- a meta-analysis. Lupus 1997;
6: 467-473.
[Abstract]
18. Wahl DG, Guillemin F, de Maistre E et al. Meta-analysis
of the risk of venous thrombosis in individuals with
antiphospholipid antibodies without underlying autoimmune
disease or previous thrombosis. Lupus 1998;7: 15-22.
[Abstract]
19. Matsuura E, Igarashi Y, Yasuda T, Triplett DA, Koike T.
Anticardiolipin antibodies recognize beta 2-glycoprotein I
structure altered by interacting with an oxygen modified
solid phase surface. J Exp Med 1994;179: 457-462.
Open Acces
20. Roubey RA, Eisenberg RA, Harper MF, Winfield JB.
Anticardiolipin autoantibodies recognize beta 2-glycoprotein
in the absence of phospholipids. Importance of Ag density
and bivalent binding. J Immunol 1995; 154: 954-960.
Abstract
21. Amengual O, Atsumi T, Khamashta MA, Koike T, Hughes GRV.
Specificity of ELISA for antibody to beta 2-glycoprotein I
in patients with antiphospolipid syndrome. Br J Rheumatol
1996;35: 1239-43.
Full Text
22. Cabral AR, Amigo MC, Cabiedes J, Alarcon Segovia D. The
antiphospholipid/cofactor syndrome: a primary variant with
antibodies to β 2 glycoprotein I but no antibodies
detectable in standard antiphospholpid assay. Am J Med
1996;101: 472-481.
23. Galli M, Luciani D, Bertolini G, Barbui T. Anti-beta 2-glycoprotein
I, antiprothombin antibodies, and the risk of thrombosis in
the antiphospholipid syndrome. Blood 2003; 102: 2717-23.
Full Text
24. Bertolaccini ML, Atsumi T, Koike T, Hughes GR, Khamashta
MA. Antiprothrombin antibodies detected in two different
assays systems. Prevalence and clinical significance in
systemic lupus erythematosus. Thromb Haemost 2005; 93:
289-297.
Abstract
25. Bertolaccini ML, Gomez S, Pareja JF et al.
Antiphospholipid Antibody tests: spreading the net. Ann
Rheum Dis 2005;64:1639-43.Open
Access
26. Meroni PL. Pathogenesis of the antiphospholipid syndrome.
An additional example of the mosaic of autoimmunity. Journal
of autoimmunity 2008;30:99-103.
Abstract
27. Branch DW, Rodgers GM. Induction of endothelial cell
tissue factor activity by sera from patients with
antiphospholipid syndrome: a possible mechanism of
thrombosis. Am J of Obstetrics & Gynecology 1993; 168:
206-10.Abstract
28. Amengual O, Atsumi T, Khamashta MA, Hughes GRV. The role
of the tissue factor pathway in the hypercoagulable state in
patients with the antiphospholipid syndrome. Thrombosis &
Haemostasis 1998; 79: 276-81.
29. Malia RG, Kitchen S, Greaves M, et al. Inhibition of
activated protein C and its cofactor protein S by
antiphospholipid antibodies. Br J Haematol 1990;76 : 101-7.
30. Atsumi T, Khamashta MA, Amengual O, et al. Binding of
anticardiolipin antibodies to protein C via beta-2-glyprotein
I (beta-2GPI): A possible mechanism in the inhibitory effect
of antiphospholipid antibodies on the protein C system.
Clinical & Experimental Immunology 1998;112: 325-33.
31. Atsumi T, Khamashta MA, Andujar C, et al. Elevated
plasma lipoprotein(a) level its association with impaired
fibrinolysis in patients with antiphospholipid syndrome. J
Rheumatol 1998;25: 69-73.
32. Lopez Pedrera Ch, Buendìa P, Aguirre MA, et al.
Antiphospholipid syndrome and tissue factor: a thrombotic
couple. Lupus 2006; 15: 161-6.
33. Hill MB, Phipps JL, Malia RG, et al. Characterization
and specificity of anti-endothelial cell membrane antibodies
and their relationship to thrombosis in primary
antiphospholipid syndrome. Clinical & Experimental
Immunology 1995;102:368-72.
34. Pierangeli SS, Colden-Stanfield M, Liu X, et al.
Antiphospholipid antibodies from antiphospholipid syndrome
patients activate endothelial cells in vitro and in vivo.
Circulation 1999;99: 1997-2002.
35. Branch DW, Dudley DJ, Mitchell MD, et al. Immunoglobulin
G fractions from patients with antiphospholipid antibodies
cause fetal death in BALB/c mice: A model of autoimmune
fetal loss. Am J of Obstetrics Gynecology 1990; 163: 210-6.
36. Chamley LW, Duncalf AM, Mitchell MD, et al. Action of
anticardiolipin and antibodies to beta-2-glycoprotein I on
trophoblast proliferation as a mechanism for fetal death.
Lancet 1998;352: 1037-8.
37. Girardi G, Redecha P, Salmon JE. Heparin prevents anti-phospholipid
antibody –induced fetal loss by inhibiting complement
activation. Nat Med 2004;10: 1222-6.
38. Mintz G, Acevedo Vazquez E, Gutierrez Espinosa G et al.
Renal vein thrombosis and inferior vena cava thrombosis in
systemic lupus erythematosus. Arthritis Rheum 1984;
27:539-544.
39. Brucato A, Baudo F, Barberis M, et al. Pulmonary
hypertension secondary to thrombosis of the pulmonary
vessels in a patient with the primary antiphospholipid
syndrome. J Rheumatol 1994; 21:942-944.
40. Day SM, Rosenzweig BP, Kronzon I. Transesophageal
echocardiographic diagnosis of right atrial thrombi
associated with the antiphospholipid syndrome. J Am Soc
Echocardiogr 1995;8:937-940.
41. Asherson RA, Higenbottam TW, Dinh Xuan AT, et al.
Pulmonary hypertension in a lupus clinic: experience with
twenty-four patients. J Rheumatol 1990; 17:1292-1298.
42. Vianna JL, Khamashta MA, Ordi-Ros J, et al. Comparison
of the primary and secondary antiphospholipid syndrome: a
European multicenter study of 114 patients. Am J Med
1994;96:3-9.
43. Allogen H, Abbott R. Cerebral Venous Sinus Thrombosis.
Review. Postgrad Med J 2000;76:12-15.
44. Christopher R, Nagaraja D, et al. Anticardiolipin
antibodies: a study in cerebral venous Thrombosis. Acta
Neurol Scand 1999;Feb:99 (2)121-4.
45. Pelletier S, Landi B, Piette JC et al. Antiphospholipid
syndrome as the second cause of non-tumorous Budd-Chiari
syndrome. J Hepatol 1994; 21:76-80.
46. Hoon Jung K, Kwon Hyun H, Kwon HA Y, et al. CT features
of abdominal manifestations of primary antiphospholipid
syndrome. J C Ass Tomography 1999;23:678-683.
47. Asherson RA, Khamashta MA, Hughes GRV. The hepatic
complications of the antiphospholipid antibodies. Clin Exp
Rheum 1991;9: 341-344.
48. Barak N, Orion Y, Scheneider M et al. Hepatic
involvement in antiphospholipid syndrome. J Gastroen Hep
1999;14: 1124-1128.
49. Asherson RA, Hughes GRV. Hypoadrenalism, Addison's
disease and antiphospholipid antibodies. J Rheumatol 1991;
18:1-3.
50. Arnason JA, Graziano FM. Adrenal insufficiency in the
antiphospholipid syndrome. Semin Arthritis Rheum
1995;25:109-116.
51. Andre M, Aumaitre O, Piette JC, et al. . Hypopituitarism
in a woman with severe primary antiphospholipid syndrome.
Ann Rheum Dis 1998;57:2578.
52. Pandolfi C, Gianini A, Fregoni V, et al. Hypopituitarism
and antiphospholipid syndrome. Minerva Endocrinol
1997;22:103-5.
53. Shah NM, Khamashta MA, Atsumi T, et al. Outcome of
patients with anticardiolipin antibodies: a 10 year of
follow up of 52 patients. Lupus 1998;7:3-6.
54. Krnic-Barrie S, Reister O’Connor C, Looney SW, et al. A
retrospective review of 61 patients with antiphospholipid
syndrome. Arch Intern Med 1997; 157:2101-2108.
55. Levine SR, Brey RL, Sawaya KL, et al. Recurrent Stroke
and thrombo-occlusive events in antiphospholipid syndrome.
Ann Neurol 1995;38:119-124.
56. Coull BM, Levine SR, Brey RL. The role of
antiphospholipid antibodies and stroke. Neurol Clin
1992;10:125-143.
57. Asherson RA, Khamashta MA, Gil A et al. Cerebrovascular
disease and antiphospholpid antibodies in systemic lupus
erythematosus, lupus-like disease and the primary
antiphospholipid syndrome. Am J Med 1989; 86:391-399.
58. Stimmler MM, Coletti PM, Quismorio FP. Magnetic
resonance imaging of the brain in neuropsyquiatric systemic
lupus erythematosus. Sem Arthritis Rheum 1993; 22:335-349.
59. Khamashta MA, Cervera R, Asherson et al. Association of
antibodies against phospholipids with heart valve disease in
systemic lupus erythematosus. Lancet 1990; 335:1541-1544.
60. Nesher G, Ilany J, Rosenman D, Abraham AS. Valvular
disfunction in antiphospholipid syndrome: prevalence,
clinical features and treatment. Semin Arthritis Rheum
1997;27:27-35.
61. Nihoyannopoulos P, Gomez PM, Joshi J, et al. Cardiac
abnormalities in systemic lupus erythematosus. Circulation
1990; 81:369-375.
62. Cervera R, Khamashta MA, Font J et al. High prevalence
of significant heart valve lesions in patients with the
primary antiphospholipid syndrome. Lupus 1991; 1:43-47.
63. Galve E, Ordi J, Barquinero J, et al. Valvular heart
disease in primary antiphospholipid syndrome. Ann Intern Med
1992; 116:293-298.
64. Amital H, Langevitz P, Levy Y, et al. Valvular
deposition of antiphospholipid antibodies in the
antiphospholipid syndrome: a clue to the origin of the
disease. Clin Exp Rheum 1999;17:99-102.
65. Garcìa Torres R, Amigo MC , De la Rossa A Moron A, Reyes
PA. Valvular Heart disease in primary antiphospholipid
syndrome: Clinical and morphological findings. Lupus
1996;5:56-61.
66. Lubbe WF, Asherson RA. Intracardiac Thrombus in systemic
lupus erythematosus associated with lupus anticoagulant.
Arthritis Rheum 1988;31:1453-1454.
67. Brown JH, Doherty CC, Allan DC, et al. Fatal cardiac
failure due to myocardial microthrombi in systemic lupus
erythematosus. Br Med J 1989;298:525.
68. Murphy JJ, Leach IA. Findings at necropsy in the heart
of a patient with anticardiolipin syndrome. Br Heart J
1989;62:61-64.
69. Hasnie AM, Stoddard MF, Gleason CB, et al. Diastolic
dysfunction is a feature of the antiphospholipid syndrome.
Am Heart J 1995;129:1009-13.
70. Coudray N, De Zuttere D, Bletry O et al. M mode and
Doppler echocardiographic assessment of left ventricular
diastolic function in primary antiphospholipid syndrome. Br
Heart J 1995;74:531-535.
71. Nochy D, Daugas E, Droz D, et al. The intrarenal
vascular lesions associated with primary antiphospholipid
syndrome. J Am Soc Nephrol 10:507-518; 1999.
72. Ostuni PA, Lazzarin P, Pengo V, et al. Renal artery
thrombosis and hypertension in a 13 year-old girl with
antiphospholipid syndrome. Ann Rheum Dis 49: 184-187; 1990.
73. Sangle S, D´Cruz D, Abbs IC, et al. Renal artery
stenosis in hypertensive patients with antiphospholipid
(Hughes) syndrome: outcome following anticoagulation.
Rheumatology 44:372-377; 2005.
74. Sangle S, D´Cruz D, Wajanat J, et al. Renal artery
stenosis in antiphospholipid syndrome and hypertension. Ann
Rheum Dis 62: 999-1002; 2003.
75. Asheron RA, Cervera R, Piette J-C, et al. Catastrophic
antiphospholipid syndrome: Clinical and laboratory features
of 50 patients. Medicine 1998;77:195-207.
76. Cervera R, Bucciarelli S, Espinosa G, et al.
Catastrophic Antiphospholipid syndrome: lessons from the
“CAPS Registry”-a tribute to the late Josep Font. Ann NY
Acad Sci 2007; 1108:448-56.
77. Khamashta MA, Machin SJ. Hematological immune cytopenias
and antiphospholipid antibodies. In: Phospholipid binding
antibodies (ed EN Harris, T Exner, GRV Hughes and RA
Asherson) 1991; pp:247-254. CRC Press, Boca Raton, Florida.
78. Uthman I, Godeau B, Taher A et al. The hematologic
manifestation of the antiphospholipid syndrome. Blood Rev
2008; 14.
79. Finazzi G. The Italian Registry of antiphospholipid
antibodies. Haematologica 1997;82:101-105.
80. Lechner K, Pabinger-Fasching I. Lupus anticoagulants and
Thrombosis. A study of 25 cases and review of the literature.
Haemostasis 1985;15:254-262.
81. Cuadrado MJ, MJ, Mujic F, Muñoz E, et al.
Trombocytopenia in the antiphospholipid syndrome. Ann Rheum
Dis 1997;56:194-196.
82. Asherson RA, Khamashta MA, Ordi-Ros J, et al. The
primary antiphospholipid syndrome: major clinical and
serological features. Medicine (Baltimore) 1989;68:366-374.
83. Moore J, Ma DD Concannon A. Non Malignant bone marrow
necrosis. A report of two cases. Pathology 1998;30: 318-320.
84. Bulvick S, Aronson I, Ress S, Jacobs P. Extensive bone
marrow necrosis associated with antiphospholipid antibodies.
Am J Med 1995;98:572-574.
85. Herranz MT, Rivier G, Khamashta MA, Blaser KU, Hughes
GRV. Association between antiphospholipid antibodies and
epilepsy in patients with systemic lupus erythematosus.
Arthritis Rheum 1994; 37:568-571.
86. Asherson RA, Derksen RWHM, Harris EN, et al. Chorea in
systemic lupus and lupus-like disease: association with
antiphospholpid antibodies. Sem Arthritis Rheum 1987;
16:253-259.
87. Cervera R, Asherson R, Font J, et al. Chorea in the
antiphospholipid Syndrome. Medicine 1997;76: 203-212.
88. Herranz MT, Rivier G, Khamashta MA, et al. Association
between antiphospholipid antibodies and epilepsy in patients
with systemic lupus erythematosus. Arthritis Rheum 1994
;37(4): 568-571.
89. Alarcon-Segovia D, Deleze M, Oria CV, Sánchez-Guerrero
J, Gómez-Pacheco L. Antiphospholipid antibodies and the
antiphospholipid syndrome in systemic lupus erythematosus: a
prospective analysis of 500 consecutive patients. Medicine
(Baltimore) 1989; 68:353-365.
90. Lavalle C, Pizarro S, Drenkard C. Transverse Myelitis:
Manifestation of SLE associated with antiphospholipid
antibodies. J Rheumatol 1990;17:34-37.
91. Sanna G, D`Cruz D, Cuadrado MJ. Cerebral manifestations
in the antiphospholipid (Hughes) syndrome. Rheum Dis Clin
North Am 2006; 72:465-90.
92. Chapman J, Rand JH, Brey RL, et al. Non stroke
neurological syndromes associated with antiphospholipid
antibodies: evaluation of clinical and experimental studies.
Lupus 2003;12: 514-7.
93. Cuadrado MJ, Khamashta MA, Ballesteros A, Godfrey T,
Simon MJ, Hughes GRV. Can neurologic manifestations of
Hughes (Antiphospholipid Syndrome) can be distinguished from
Multiple Sclerosis? Medicine 2000;79:57-68.
94. Montalban J, Cervera R, Font J et al. Lack of
association between anticardiolipin antibodies and migraine
in systemic lupus erythematosus. Neurology 1992; 42:681-682.
95. Tsakiris DA, Kappos L, Reber G et al. Lack of
association between antiphospholipid antibodies and migraine.
Thromb Haemost 1993; 69:415-417.
96. Asherson RA, Mayou SC, Merry P, Black MM, Hughes GRV.
The spectrum of livedo reticularis and anticardiolipin
antibodies. Br J Dermatol 1989; 120:215-221.
97. Levine SR, Langer SI, Albers JW, et al. Sneddon’s
syndrome: An antiphospholipid antibody syndrome? Neurology
1988;38:798-800.
98. Alegre VA, Winkelmann RK, Gastineau DA. Cutaneous
Thrombosis , cerebrovascular thrombosis and lupus
anticoagulant in the Sneddon syndrome: report of 10 cases.
Int J Dermatol 1990;29:45-9.
99. Toubi E, Krause I, Fraser A, et al. Livedo reticularis
is a marker of predicting multisystem thrombosis in
antiphospholipid syndrome. Clin Exp Rheumatol 2005; 23:
499-504.
100. Alegre VA, Gastineau DA, Winkelman RK. Skin lesions
associated with circulating lupus anticoagulant. Br J
Dermatol 1989;120:419-429.
101. Gibson GE, Daniel Su WP, Pittelkow MR. Antiphospholipid
Syndrome and the skin. J Am Acad Dermatol 1997;36:970-982.
102. Ishikawa O, Takahashi A, Tamura A, Miyachi Y. Cutaneous
papules and nodules in the diagnosis of antiphospholipid
syndrome. Br J Dermatol 1999;140: 725-729.
103. Asherson RA. Subungueal splinter haemorrages.: A new
sign of the antiphospholipid coagulopathy? Ann Rheum Dis
1990; 49:268-71
104. Stephanson E Niemi KM. Antiphospholipid antibodies and
anetoderma: Are they associated ? Dermatology
1995;191:204-209.
105. Oshiro BT, Silver RM, Scott JR, et al. Antiphospholipid
Antibodies and fetal death. Obstet Gynecol 1996;87:489-93.
106. Lockwood CJ, Romero R, Feinberg RF et al. The
prevalence and biologic significance of lupus anticogulant
and anticardiolipin antibodies in a general obstetric
population. Am J Obstet Gynecol 1989; 161:369-373.
107. Harris EN, Spinnato JA. Should anticardiolipin tests be
performed in otherwise healthy pregnant women? Am J Obstet
Gynecol 1991; 165:1272-1277.
108. Lockshin MD, Qamar T, Druzin ML, Goei S. Antibody to
cardiolipin, lupus anticogulant, and fetal death. J
Rheumatol 1987; 14:259-262.
109. Buchanan NMM, Khamashta MA, Morton KE, Kerslake S,
Baguley EA, Hughes GRV. A study of 100 high risk lupus
pregnancies. Am J Reprod Immunol 1992; 28:192-194.
110. Branch DW, Silver RM, Blackwell JL, Reading JC, Scott
JR. Outcome of treated pregnancies in women with
antiphospholipid syndrome: an update of the Utah experience.
Obst Gynecol 1992; 80:614-620.
111. Balasch J, Font J. Antiphospholipid antibody testing in
patients with pregnancy loss. Lupus 1994; 3:429-431.

|