|
TRABAJO ORIGINAL
Rol de las mitocondrias y de
las especie reactivas de oxigeno en la pro-gresion de la
insuficiencia cardiaca
Role of mitochondria and reactive oxygen species in
the progression of heart failure
Gustavo Guzmán
Mentesana; Alejandra Baez; Roque Córdoba; Ricardo Domínguez;
Silvina Lo Presti; Walter Rivarola; Patrícia Pons; Ricardo
Fretes; Patrícia Paglini-Oliva.
Revista Facultad de Ciencias
Medicas 2010; 67(4): 150-158
Cátedras de Física Biomédica,
Histología y Microscopía Electrónica, Universidad Nacional
de Córdoba, Sanatorio Allende y Clínica Aconcagua, Córdoba ,
Argentina
Introducción
La compresión de los mecanismos que conducen a la
insuficiencia cardiaca, es uno de los mayores retos de la
cardiología actual. Interpretar las vías neurohormonales y
como éstas interactúan a nivel celular y molecular, es el
comienzo para alcanzar este gran objetivo. ()
El corazón es un órgano con un gran consumo de energía por
ello es altamente oxidativo; este vital recurso es aportado
por las mitocondrias que representan aproximadamente el 30%
del volumen total de los cardiomiocitos y le proveen el 90%
de la energía que ello requieren(). Tal actividad oxidativa
las hacen blancos de las especie reactivas del oxigeno
(ROS). ().
Involucrado en la vía de síntesis del ROS, moléculas
inestables, se encuentran la enzima oxido nitro sintasa
(NOS), de las cuales se reconocen dos isoformas: las
constitutivas (NOS endotelial eNOS, NOS neural nNOS) y las
inducibles (iNOS)
(1).
El rol principal de las NOS es la síntesis del oxido nítrico
(NO) el cual participa de forma importante en la
funcionamiento cardiaco provocando efectos vasculares como
vasodilatación y no vasculares como su intervención en la
regulación de la producción de energía en la cadena
respiratoria
2 .
La mayor parte del oxígeno utilizado por los miocitos es
reducido a agua por vía del complejo IV mitocondrial. En
esta reducción del O2 pueden producir radicales libres
dependientes del oxigeno (ROS) como ser aniones superóxido
(O2-), peróxido de hidrogeno (H2O2) y radicales hidroxilo
(OH); por otro lado la reacción del anión superόxido origina
los peroxinitritos (ONOO-) llamado especies reactivas del
nitrito (ERN) que son altamente citotóxicos ya que causan la
oxidación de los fosfolípidos de membrana, proteínas y
material genético (3),
estos ha sido implicados en una amplia gama de condiciones
patológicas incluyendo lesión de isquemia, enfermedades
neurodegenerativas y el envejecimiento. En virtud de los
efectos tóxicos que estas biomoleculas producen a nivel
celular, existen enzimas (basureros) como: superóxido
dismutasa (SOD), glutation peroxidasa (GSHPx), catalasa y
otros antioxidantes no enzimáticos capaz de prevenir de los
efectos tóxicos de los ROS. Sin embargo si la producción de
ROS es excesiva el estrés oxidativo puede tener un efecto
perjudicial sobre la función y la integridad de los tejidos
biológicos
(4).
Estudios experimentales y clínicos han sugerido que el
estrés oxidativo es uno de los principales parti-cipantes en
la remodelación y fracaso miocárdico, estos hechos se
asocian a que los corazones con in-suficiencia cardiaca
congestiva (ICC) presentan un incremento de ROS y reducción
de enzimas anti-oxidantes, además este aumento crónico de
radicales dependiente del oxígeno en la mitocondria puede
conducir a un ciclo catastrófico de daños en el ADN
mitocondrial (ADNmt), así como la disminución funcional del
miocardio mediada a través de una lesión celular. Especies
reactivas de oxígeno pueden inducir hipertrofia del miocito,
apoptosis y fibrosis intersticial mediante la activación de
metaloproteinasas de matriz. Estos eventos celulares juegan
un papel importante en el desarrollo y la progresión de la
remodelación miocárdica
(5).
También se han hallado en músculo cardiaco alteraciones a
nivel ultraestructural, principalmente en la estructura y
función mitocondrial(6-7);
en miocardiopatias hipertrofias y dilatadas se identifican
defectos en la cadena respiratoria y en la fosforilación
oxidativa (8,9),
así como mutaciones en el citocromo c y deleciones del DNA
mitocondrial, cuya relación con la cardiopatía aún no ha
sido explicada
(7, 9, 10);
los defectos de conducción y las arritmias se han detectado
en pacientes que también tenían alteraciones en la
oxidación.
Por otro lado se han postulados que las alteraciones
mitocondriales para el músculo cardiaco serian acompañados
de daños similares en al misma organela del músculo
esquelético(11).
Es por ello que el objetivo del presente trabajo fue
establecer la influencia del ROS (medida por el ion
peroxidonitrito y la enzima iNOS) sobre la estructura y
función mitocondrial de músculo cardiaco y es-quelético en
pacientes con insuficiencia cardiaca congestiva (ICC) grado
III y IV según New York Heart Association (NYHA), hecho que
contribuiría por un lado a la mejor comprensión de los
factores involucrados en la progresión de la ICC y por otro
conocer el estado celular del corazón mediante una biopsia
de cualquier músculo estriado.
Materiales y Métodos
Pacientes
Fueron incluidos en este estudió aquellos pacientes que
fueron sometidos a cirugía cardiovascular por diferentes
motivos; a ellos se les confeccionó una historia clínica con
datos epidemiológicos, metabólicos, hábitos tóxicos, signos
y síntomas de los diferentes grado de insuficiencia cardiaca
congestiva como así también fármacos utilizados para su
tratamiento. De ellos se obtuvo una muestra de ventrículo
izquierdo y de músculo esquelético (músculo pectoral mayor)
de 1 mm2.
Criterios de inclusión
Pacientes control (n= 7)
1. Paciente menores 40 años, de ambos sexos.
2. Fracción de Eyección > 60%.
3. Sin enfermedad reumatológicas.
4. Con comunicación Interauricular como diagnostico previo a
la cirugía.
Pacientes con miocardiopatías (n=18)
1. Pacientes mayores de 40 años, ambos sexos.
2. Enfermedad Cardiovascular Secundaria.
3. ICC grado III – IV según la NYHA.
4. Sin enfermedades reumatológicas o inmunológicas.
Preparación del material para microscopia electrónica.
Las biopsias de miocardio y músculo esquelético de los
pectorales, se fijaron en solución de Karnovsky compuesto de
una mezcla de formaldehído al 4 % y glutaraldehído 1.5% en
el tampón cacodilato 0.1 M durante un período mínimo de 2 hs
a temperatura ambiente. El glutaraldehído al 25% fue
previamente purificado por destilación y el formaldehído
preparado por depolimerización de paraformaldehído en
solución acuosa a 80º y con ligera alcalinización. A
continuación, el tejido fue lavado 3 veces en tampón
cacodilato y tratado con tetróxido de osmio al 1% en la
misma solución tampón a temperatura ambiente durante 1-2 hs.
Luego, el material fue deshidratado en soluciones acuosas de
acetona de graduaciones crecientes (50%, 70% y 90%) durante
5 minutos en cada uno de ellas y el proceso fue completado
con 3 pasajes, de 15 minutos cada uno, en acetona 100%
destilada y deshidratada sobre tamiz molecular Nº 3 (Merck).
La inclusión de las células se realizó en una mezcla de
resina epóxicas compuesta de: Araldita 506 (48.5%),
Anhidrido dodecenilsuccínico (DDSA) (48.5%), Dibutilftalato
(DBP) (0.5%), Acelerador dimetilaminobenceno (BDMA) (2.5%).
Estudios ultraestructurales
El examen se llevó a cabo con microscopio electrónico de
transmisión Zeiss. La randomización fo-tográfica no fue
completa ya que fueron excluidas aquellas que presentaron
artefacto de técnicas. La decisión de excluirlas fue tomada
por dos observadores expertos. El área fotografiada fue
magnificada a 10000 y 27800 X y magnificaciones mayores
fueron utilizadas para detalles específicos. De la biopsia
de 1 mm2 se le realizaron los cortes y se eligieron 10
cortes seriados a los que se tomaron en promedio 2.5
micrografías por corte. Los cambios estructurales observados
se analizaron con el programa axion-vision 4.6,
referenciados a una escala de 1 μm, a través del ello se
determino el área promedio ocupadas por las mitocondrias y
diámetro promedios de las mitocondrias en los diferentes
tejidos.
Aislamiento mitochondrial
La biopsia de músculo esquelético y cardiaco fueron lavados
y suspendidos en un buffer a 4ºC inme-diatamente
homogeneizado. Los homogenatos fueron centrifugados a 1500
g, 4ºC por 20 min y el so-brenadante transferido a un nuevo
tubo, se centrifugó nuevamente a 10000 g 4ºC por 5 m. El
pellet obtenido se lavó con el buffer y centrifugó a 10000 g
4ºC por 10 m dos veces. El pellet rico en mitocondrias se
resuspendió en buffer (relación tejido/buffer: 1/1) y las
alícuotas se guardaron a –80ºC. La concentración de
proteínas se midió por el método de Bradford. Esta fracción
sirvió para determinar:
Estudio de actividad functional
Actividad de la cadena respiratoria a través del
complejo III (ubiquinona-citocromo c oxidoreductasa): las
mitocondrias fueron suspendidas en 50 mM Tris-HCL de buffer,
pH 7.4 que contenía 1mM EDTA, 250 mM sacarosa, 2mM KCN y 50
µM citocromo c oxidado. Después de la adición de 80µM de DB
(DBH2) reducido, se midió la reducción de citocromo c a 550
nm (ε 19.0 mM-1 cm-1). Los resultados de la actividad
enzimática se expresaron en µM de ubiquinona min-1 mg prot.
Estudio de inmunohistoquimica
La expresión proteica de NOSi se analizó usando anticuerpos
Ig G policlonal específicos contra el ex-tremo amino
terminal de iNOS (Santa Cruz Biotechnology Company, Inc.
Sc8310).
Los cortes se desparafinaron con xilol y rehidrataron en una
serie sucesiva de alcoholes (100º, 96º, 70º, 50º) hasta
llegar a PBS 1X pH 7,3. La actividad peroxidasa endógena se
bloqueo con peróxido de hidrógeno 3% en PBS por 15 minutos.
Los sitios de unión inespecífica de las inmunoglobulinas
fueron bloqueados por un preincubado de 15 minutos con 5% de
SBF (GIBCO BRL, Grand Island, N. Y.) diluido en PBS, y se
lavó 3 veces con PBS.
Se incubaron los cortes con el anticuerpo primario, anti IgG
de humano contra NOSi obtenido en conejo en una dilución
1:200 durante la noche a 4ºC.
Luego de descartar el anticuerpo primario y secar alrededor
del corte se coloco el anticuerpo secundario, anti-IgG
Biotinilado en PBS por 30 minutos a temperatura ambiente. Se
lavó tres veces con PBS y se secó.
Se coloco la Peroxidasa conjugada con Estreptavidina por 20
minutos a temperatura ambiente, y luego de lavar y secar se
incubo con la solución del sustrato de la Peroxidasa y el
cromógeno, DAB (3,3’diaminobencidina) en Peróxido de
Hidrógeno 0,3% H2O2 en Buffer Tris 0,05M pH 7,6 controlan-do
en microscopio hasta obtener coloración, aproximadamente 1
minuto.
Análisis de la producción de Nitrotirosina
Se realizó en cortes de 5µm montados en vidrios con gelatina
la inmunomarcación para la nitrotirosina como un índice de
estrés oxidativo.
Los cortes se desparafinaron con xilol y rehidrataron en una
serie sucesiva de alcoholes (100º, 96º, 70º, 50º) hasta
llegar a PBS 1X pH 7,3. La actividad peroxidasa endógena se
bloqueo con peróxido de hidrógeno 3% en PBS por 15 minutos.
Los sitios de unión inespecífica de las inmunoglobulinas
fueron bloqueados por un preincubado de 1 hora con 20% de
SBF (GIBCO BRL, Grand Island, N. Y.) diluido en PBS.
Se incubaron los cortes con el anticuerpo primario:
anticuerpo monoclonal anti – nitrotirosina de conejo 1/100
(24 horas en cámara húmeda a 4 °C )
Luego de descartar el anticuerpo primario lavar con PBS, se
secó alrededor del corte se coloco el anti-cuerpo
secundario, multilinker, anti-IgG Biotinilado en PBS por 1
hora a temperatura ambiente en cámara húmeda. Se lavó tres
veces con PBS y se secó.
Se colocó la Peroxidasa conjugada con Estreptavidina por 20
minutos a temperatura ambiente, y luego de lavar y secar se
incubo con la solución del sustrato de la Peroxidasa y el
cromógeno, DAB (3,3’diaminobencidina) con Peróxido de
Hidrógeno 0,3 % H2O2 en Buffer Tris 0,05M pH 7,6 contro-lando
en microscopio hasta obtener coloración, aproximadamente 1
minutos.
Análisis Estadísticos
Los análisis estadísticos utilizados fueron ANOVA y Χ2,
estableciéndose diferencias significativas cuando p <0.05.
Resultados
Morfología de las mitocondrias del músculo cardiaco
Se analizaron las diferencias encontradas en las
mitocondrias aisladas de ventrículo izquierdo del grupo con
ICC grado funcional de la NYHA III – IV y se compararon con
las del grupo control.
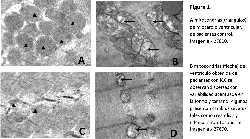
Se observó un marcado polimorfismo ultraestructurales
(figura 1) en el grupo con ICC siendo los as-pectos como
lisis y dilatación de crestas, aumento de matriz (edema) los
hallazgos mas representativos; también se observó una
reducción del área total ocupadas por las mitocondrias del
78% en los pacientes con ICC vs control 160,37µm 2±9,87;
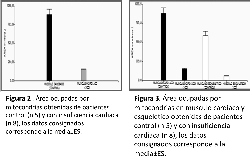
936,81µm 2±78,48 respectivamente (figura 2) p<0.0001.
Alteraciones similares se observaron en el músculo
esquelético (figura 1) con una reducción del área
mitotocondrial total en un 75% con respecto al control
90,86µm 2±6,87; 550,62µm 2±56,48 respectiva-mente p<0.0001.
|
 |
|
Es de marcar que el área ocupada por las mitocondrias en el
músculo cardiaco es notablemente mayor que el área ocupada
por dicho organoide en músculo esquelético, respetando una
relación 1,7:1 esta tendencia se mantiene tanto en el grupo
control como en el grupo con ICC (figura 3) p<0.0001.
Actividad enzimática del CIII
Se observo una reducción del 70% en la actividad del
complejo III (figura 4) en pacientes con ICC gra-do
funcional de la NYHA III – IV comparado con el control 1,9
10-2 mM ubiq.mim-1.mg prot ± 12,6; 5,79 10-2mM ubiq.mim-1.mg
prot±36,6, p<0,001.
|
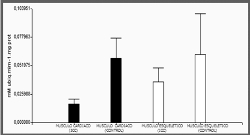 |
Figura 4.
Actividad enzimatica mitocondrial del complejo
III, en músculo cardiaco y esquelético. Obtenido
de pacientes control (n 3) y grupo con
insuficiencia cardiaca (n 6), los datos
consignados corresponde a la media±ES.
|
Análisis Inmunohistoquimico
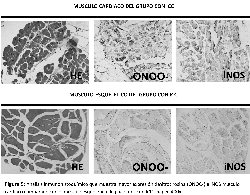
Con el objetivo de demostrar la producción de ROS se
llevaron a cabo estudios inmunohistoquímicos que demostraron
a través de la lipoperoxidacion y la presencia de la enzima
oxidonitro sintasa inducible (iNOS) una marcación intensa de
distribución difusa en más del 80% para nitrotirosina e iNOS
en el músculo cardiaco del grupo con ICC en comparación con
el grupo control (Tabla 1). La marcación para iNOS estuvo
presente en membrana y citoplasma en cambio la nitrotirosina
solo en citoplasma.
Por otro lado en el músculo esquelético de estos mismos
pacientes se observó una leve marcación difusa en todos los
casos con una distribución similar a la observada en
miocardio, no mostrando diferencia con el grupo control
(figura 5).
|
 |
Tabla 1.
Estudio inmunohistoquímicos de oxidonitro
sintasa inducible (iNOS) y nitrotirosina en
músculo cardiaco (MC) y esquelético (ME) de
pacientes con insuficiencia cardiaca
congestiva(ICC) y pacientes control
+++ intenso, ++ moderado, + leve. CITOPL.
(citoplasma) MEMB. (membrana) |
Discusión
El corazón es un tejido
altamente oxidativo y esencialmente dependiente de la
energía que aportan las mitocondrias para contraerse y
llevar a cabo otras actividades metabólicas, representan
aproximada-mente el 30% del volumen total de los
cardiomiocitos y le proveen el 90% de la energía.
La producción de energía por parte de las mitocondrias
dependen de factores genéticos del DNA mito-condrial
(12-13)
que modulan el normal funcionamiento de la actividad
enzimática de la organela y de fac-tores ambientales que
incluyen los aportes de azúcares, lípidos, proteínas y
oxígeno
(14).
Cambios en la estructura y la función de las mitocondrias se
han encontrado cada vez más asociados con las enfermedades
cardiovasculares como hipertrofia y la miocardiopatía
dilatada,(8,9),
muerte súbita, miocardiopatía isquémica, alcohólica y
miocarditis.
En el presente trabajo se observaron que los pacientes
portadores de ICC grado III-IV presentaron en musculo
cardiaco una reducción del área ocupada por las mitocondrias
del 78% con respecto al control (p<0.0001), también se
observaron en dichos organoides degeneración hidrópica,
alteraciones en mem-brana externa y crestas mitocondriales
sugieren un daño grave e irreversible de la mitocondria,
imposi-bilitando a ésta a mantener un potencial de membrana
mitocondrial efectivo debido a la perdida el gra-diente
transmembrana. La disminución en la capacidad de generar
energía se puede presuponer por una disminución en la
superficie de la membrana interna (crestas) que es el sitio
de anclaje de la cadena respiratoria. Estos sitios claves de
lesión condicionaría una alteración en la homeostasis
mitocondrial y celular, motivo por el cual se activaría las
vías apoptóticas del miocardio que conducen directa o indi-rectamente
al remodelado cardiaco
(15).
Alteraciones estructurales similares se encontraron en el
musculo esquelético de los pacientes con ICC como ser una
disminución del área ocupadas por las mitocondrias del 72%
como respecto del control (p<0.0001) patrón similar al
observado por otros autores en el modelo experimental
realizado en ratones
(16).
Este paralelismo permitiría inferir de manera relativamente
sencilla el estado celular del corazón del paciente con ICC,
mediante biopsia de cualquier músculo estriado
(17,18).
En nuestro trabajo se observo que el grupo con ICC, presento
una marcada caída en la actividad en-zimática del CIII con
respecto al control (p<0.0001). Algunos estudios han
demostrado que la cadena respiratoria mitocondrial es una
fuente de ROS y que los complejos CI y CIII son los sitios
fundamen-tales para la génesis de radicales libres
(4).
También esta alta tasa metabólica induce a la producción de
ROS
(19)
y de especies reactivas del nitrógeno RNS que en condiciones
de normalidad se encuentran en un equilibrio homeostático.
Los ROS son moléculas que en su estructura atómica presentan
un electrón de lábil unión en el orbital externo, lo que les
da una configuración espacial que genera inestabilidad. Son
extraordinariamente re-activos y de vida media corta. Estos
ROS son elaborados continuamente como productos del metabo-lismo
normal de las células y se inactivan por un conjunto de
mecanismos cuya función es equilibrar la producción de ROS y
antioxidantes para minimizar y retardar la aparición de
daños
(20).
Debido a la alta inestabilidad atómica de los ROS, pueden
sustraer un átomo de H+ dejandolo oxidado y produciendo que
determinadas moléculas pierdan su función específica en la
célula. Cuando esto ocurre en las membranas lipídicas
celulares, la oxidación de un ácido graso lo convierte en
radical de ácido graso con capacidad de oxidar a otra
molécula. Este proceso es conocido como lipoperoxidación y
permite cuantificar la concentración de los lípidos
hidroperoxidados y por ende de ROS, a través de sus
productos (ej.nitotorosina)(21).
Nuestro trabajo
observamos que los miocardiocitos expresaron una marcación
intensa del ONOO- en el 80% de caso con ICC comparado con el
control, situación que no se observo con el musculo esquelé-tico.
La formación de los ONOO- esta vinculada a la reacción del
NO intramintocondrial con el oxigeno el cual estaría
aumentado por el bloqueo del complejo IV
(22);
dicho bloqueo se observa en proceso que tiendan a aumentar
desmesuradamente el NO. La síntesis del NO se debe a las
enzimas NOS constitu-cionales e inducibles, estas ultimas
aumentan en procesos como enfermedad coronaria, diabetes,
proce-sos crónicos.
(23, 24)
La iNOS en fases iníciales de la ICC presenta función
protectora, aunque si se da lugar a un exceso las
consecuencias pueden ser fatales debido al bloqueo de la
cadena respiratoria
(25).
Otro hallazgo observado en este trabajo fue que tejido
miocardico del grupo con ICC presento una marcación intensa
(80%) de la iNOS con respecto al control, no mostrando dicha
diferencia en el mus-culo esquelético.
Entonces se observa una relación entre la mayor marcación de
la iNOS y la expresión de nitrotirosina en los miocitos,
sugiriendo que en este momento el efecto beneficioso del NO
se torna peligroso, debido al aumento crónico de radicales
libres (26),
deviniendo el fracaso energético mitocondrial y activando
diferentes vías de muertes celulares dentro del miocardio
(27, 28)
En el presente trabajo se observó una severa alteración
estructural en las mitocondrias del músculo ven-tricular
izquierdo en paciente que presentaban ICC grado III – IV con
respecto al control, asociado a una reducción de la
actividad del complejo III y una marcación aumentada de la
iNOS y nitrotirosina, observaciones que muestran una clara
relación fisiopatologíca entre ellas y permiten entender en
parte los proceso celulares y moleculares de la
insuficiencia cardiaca.
AGRADECIMIENTOS
Este trabajo se llevó a cabo con subsidios de la Secretaria
de Ciencia y Tecnología de la Universidad Nacional de
Córdoba y Ministerio de Ciencia y Tecnología de la Provincia
de Córdoba.
Bibliografía
1. Alderton WK, Cooper CE, Knowles RG. Nitric oxide
synthases: structure, funtion and inhibition. Biochem. J.
2001, 357:593.
Full Text
2. Massion PB, Feron O, Dessy C, Balligan JJ. Nitric Oxide
and cardiac function: ten years after, and continu-ing 2003,
Cir. 93: 388.
Full Text
3. J.M. McCord, Oxygen-Derived Free Radicals in Post-Ischemic
Tissue Injury, New Eng. J. Med. 1985 312:159-163.
PubMed
4. Tsutsi H. Oxidative stress in heart failure : the role of
mitochondria. Int. Med. 2006, 40, 1177-1182.
Full Text
5. Anilkumar N, Sirker A, Shah AM. Redox sensitive signaling
pathways in cardiac remodeling, hypertrophy and failure
Front Biosci. 2009 14:3168-87.
Full Text
6. Garg, N. M Front in Bioscience 2005, 10:1341-1345.
7. Marín-Garcia J, Goldenthal MJ, Ananthakrishnan R,
Pierpont MF. J Card Fail 2000, 6: 321-329.
8. Rustin P, Lebidois J, Chretien D, Bourgeron T, Piechaud
JF et al. J Pediatr 1994, 124: 224-228.
9. Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, et
al. Am J Pathol 1998, 153: 1501-1510.
10. Schon EA, Bonilla E, DiMauro S. J Bioenerg Biomembr
1997, 29: 131-149.
11. Marin- Garcia J. J Inherit Metab Dis 1997, 20: 674-680.
12. Anderson S, Bankier AT, Barrell BG, DE Brujin et al.
Nature 1981, 290: 457-465.
13. Shadel GS, Clayton DA. Annu Rev Biochem 1997, 66:
409-435.
14. Marin- Garcia J. Rev Esp Cardiol 2002, 55: 1293-1310.
15. Kumar S, Kain V, Sitasawad SL:Cardiotoxicity of
calmidazolium chloride is attributed to calcium aggrava-tion,
oxidative and nitrosative stress and apoptosis. Free Radic
Biol Med. 2009.
PubMed
16. Garg N. Mitochondrial disorders in chagasic
cardiomyopathy. Front Biosci. 2005, 10:1341-1354.
PubMed
17. Marin-Garcia J, Pi Y, Goldenthal MJ. Cardiovasc Drugs
Ther. 2006, 20: 477-49
18. Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura
K, et al. Mitochondrial DNA damage and dys-function
associated with oxidative stress in failing hearts after
myocardial infarction. Circ Res 2001;88:529-535.
Full text
19. Lenaz, G., Bovina, C., D´Aurelio, M., Fato, R.,
Formiggini, G., Genova, M., Giuliano, G., Pich, M., Paolucci,
U., Castelli, G.. Ann N. Y. Acad. Sci. 2002, 959, 199-213.
20. Halliwell B, Whiteman M: Measuring reactive species and
oxidative damage in vivo and in cell culture: how should you
do it and what do the results mean?. Br J Pharmacol, 2004,
142:231-55
PubMed
21. Le Chen, Qizhi Gong, James P. and A.A. Knowlton:
Mitochondrial OPA1, Apoptosis and Heart Failure. Car-diovasc
Res: 2009, 181v1-181.
Full
Text
22. Kuzcaya N, Weissmann N, Harrison D G, Dikalov S.
Interactions of peroxynitrite, tetrahydrobiopterin, ascor-bic
acid, and thiols. J Biol. Chem. 2003, 278: 22546.
Full Text
23. Guillermo Zalba, Fortuño Ana, Díez Javier: Oxidative
stress and atherosclerosis in early chronic kidney dis-ease.
Nephrol Dial Transplant, 2006, 21:2686-2690.
Full text
24. Liu Shang Xi, Hou Fan Fan, Guo Zhi Jian, Nagai Ryoji,
Zhang Wei Ru, Liu Zhi Qiang, Zhou Zhan Mei, Zhou Mei, Di Xie
, Wang Guo Bao, Zhang Xun: Advanced Oxidation Protein
Products Accelerate Atherosclerosis Through Promoting
Oxidative Stress and Inflammation. Arterioscler Thromb Vasc
Biol, 2006, 26:1156-1162.
Full Text
25. Cadenas E, Poderoso JJ, Antunes F, Boveris A. Analysis
of the pathways of nitric oxde utilization in mito-condria.
Free Radic 2000, Res 33:747
PubMed
26. Wen JJ, Bhatia V, Popov VL, Garg NJ. Am J Pathol. 2006,
169:1953-1964.
27. Long X, Goldenthal MJ, Wu GM, Marin-Garcia J. Mol Cell
Cardiol. 2004, 37: 63-70.
28. Viatkina G, Vandanajay B, Arpad G, Papaconstantinou
J.Biochim Biophysic 2004, 689: 162-173;.

|