|
TRABAJO DE CASUÍSTICA Y CASOS
CLÍNICOS
Xantomatosis plana difusa
asociada a tumor hematológico y neoplasia sólida. Hallazgos
de una autopsia
Didduse plane xanthomatosis associated with hematologic
disorder and solid tumos. findings of an autopsy
María Virginia Bürgesser*, Calafat Patricia*, Diller Ana*
Revista Facultad de Ciencias
Medicas 2011; 68(2): 65-69
*Servicio de Patología, Hospital
Privado de Córdoba. Córdoba, Argentina
Autor para
correspondencia:
Bürgesser, María Virginia
Dirección: Naciones Unidas 346. Barrio Parque Vélez
Sarsfield. Córdoba, Argentina. CP: X5016KEH
Teléfono: 0351-4688829; Fax: 0351-4688826
Email:
virburgesser@gmail.com
Introducción
La xantomatosis plana difusa es
una entidad poco frecuente, de etiología desconocida,
clasificada como un subtipo de histiocitosis no Langerhans.
Suele manifestarse por placas pardoamarillentas que se
presentan en cualquier zona de la superficie corporal. Puede
presentarse, además, con lesiones mucosas, sobre todo en
tracto respiratorio superior, y con lesiones extracutáneas
como compromiso de hipófisis y desarrollo de diabetes
insípida en un 40%. Su asociación con trastornos
hematológicos, en especial con gammapatías monoclonales y
mieloma múltiple, ha sido descripta en un 20% de los casos.
La coexistencia con neoplasias sólidas se ha reportado,
aunque con escasa frecuencia.
Se presenta un caso de autopsia destacando el diagnóstico de
xantomatosis plana difusa con extenso compromiso de órganos
internos, asociada a dos neoplasias, un trastorno
hematológico y un tumor sólido.
Reporte de caso
Paciente de sexo femenino de 78 años con antecedentes de
gammapatía monoclonal de significado incierto (MGUS) con
pico monoclonal kappa desde 1994 con dos biopsias de médula
ósea que mostraban un leve aumento de células plasmáticas en
los años 1994 y 2002. Presentaba anemia de proceso crónico
con hemoglobina de 11 a 12gr/dl. Además, tenía diagnóstico
clínico de xantomatosis plana difusa desde 1995 con
múltiples lesiones pardoamarillentas diseminadas
características. En su evolución se le realizaron tres
biopsias de piel (de brazo derecho, de dorso y de abdomen)
sin hallazgos evidentes de xantomatosis. Padecía demencia
senil de 5 años de evolución con dependencia para las
actividades diarias en el último año.
Fue traída por sus familiares a la consulta de urgencias por
deterioro del estado general. Al examen físico se evidenció
síndrome consuntivo, con severa disminución de masas grasa y
muscular, palidez cutáneo-mucosa, sin masas palpables en
abdomen, ni hepatoesplenomegalia. Se realizó laboratorio que
constató anemia con franca disminución de los valores de
hemoglobina (Hb: 5,8 gr/dl – Hto: 21,5%; VCM: 64,4 fl; HCM:
17,4 pg; CHCM: 27 gr/dl; RDW: 18.8%) con ligera elevación de
GB (13.900/ml – neutrófilos segmentados: 61% linfocitos:
24%). Se realizó ecografía abdominal que mostró
engrosamiento focal de pared de colon proximal. Una
radiografía de tórax reveló infiltrado alveolar en campos
inferiores y campo medio de pulmón izquierdo. Se decidió
internación en sala común y realización de medidas
paliativas. Al cuarto día de internación presento fiebre y
secreciones respiratorias, constatándose el óbito a las 24
horas. Se solicitó autopsia sin sistema nervioso central.
Los hallazgos identificados como enfermedad de base fueron:
xantomatosis plana difusa asociada a enfermedad hematológica
con compromiso extenso de piel, en forma diseminada, con
mayor número de lesiones en cabeza, cuello y dorso. A la
apertura de la cavidad toracoabdominal, so observaron
múltiples adherencias pleurales a pared torácica y
pericardio. Además la serosa peritoneal y el mesenterio
exhibían un aspecto despulido. Al examen microscópico se
reconoció una infiltración difusa de dermis superficial y
profunda por células medianas a grandes, de citoplasma
amplio y vacuolado con disposición perivascular, perianexial
y perineural (figuras 1 y 2). Las mismas fueron positivas
para CD68 y negativas para S100. Dicha infiltración se
extendía a la pared del tracto gastrointestinal (figura 3),
a la cápsula de Gleason, pared de aurícula derecha, válvulas
cardíacas derechas y tejido fibroadiposo pericoronario, así
como a pericardio, pleura, peritoneo, mucosa vesical y
médula ósea.
|
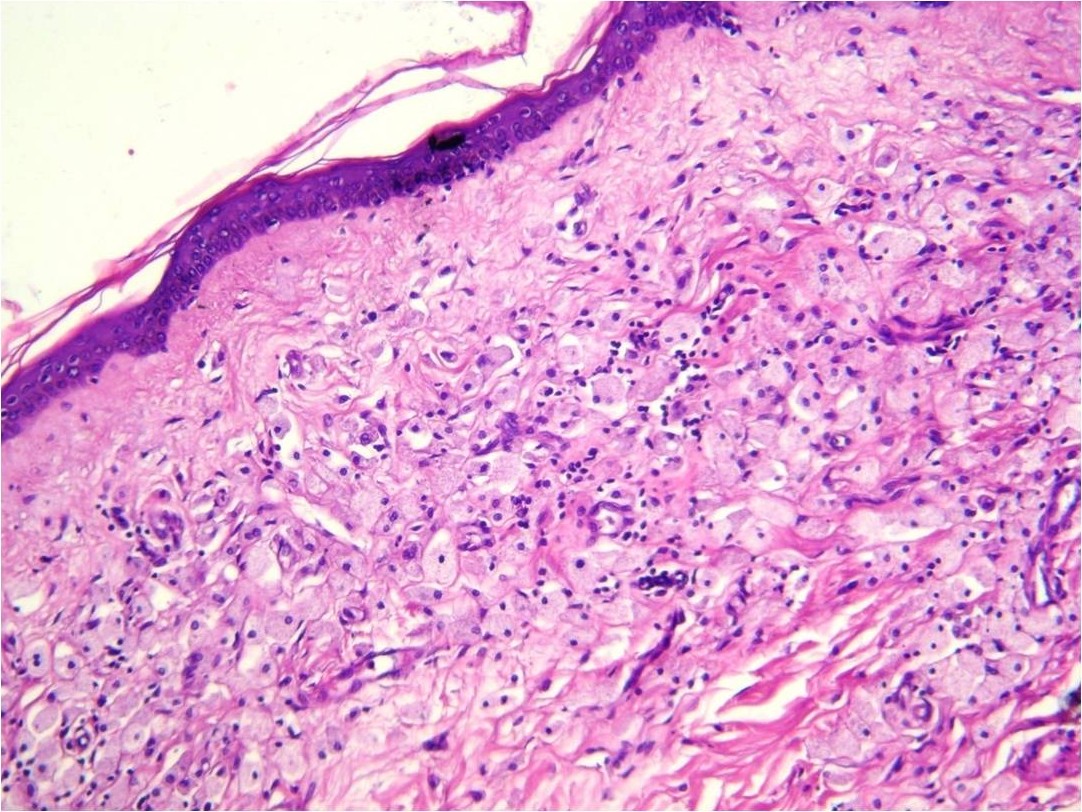 |
Figura 1: H/E
20x: infiltración difusa de dermis superficial
por células xantomatosas |
|
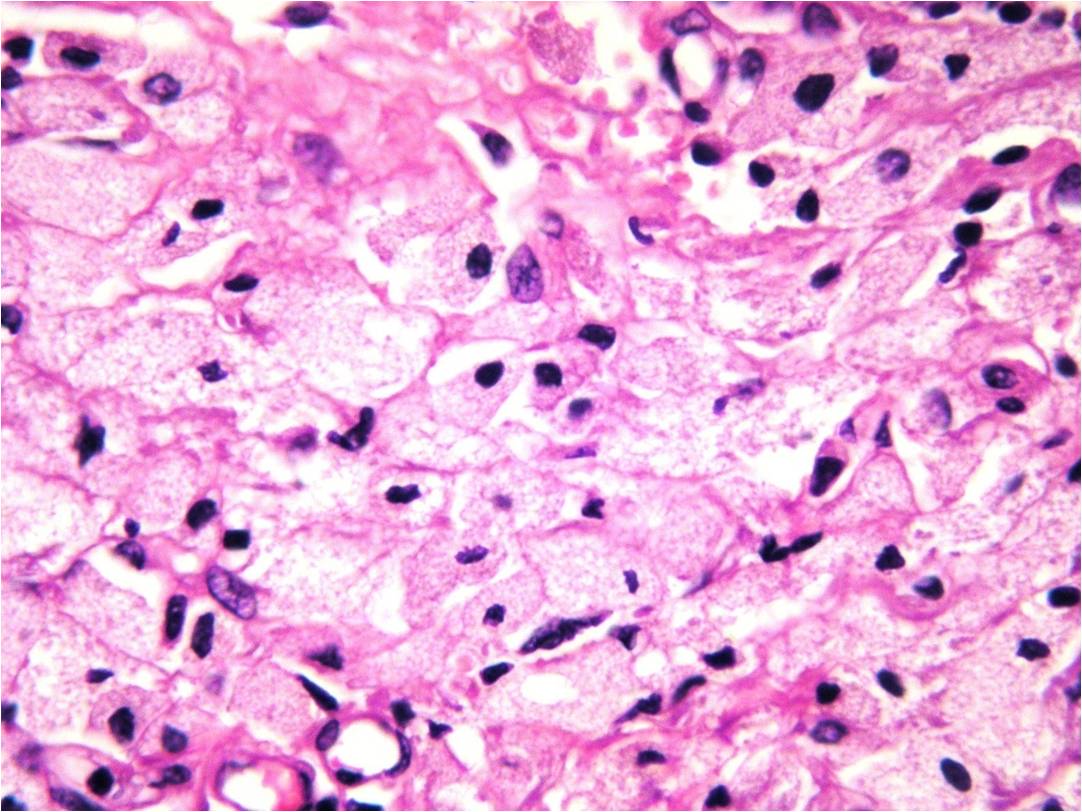 |
Figura 2: H/E
40x: células medianas de citoplasma amplio
vacuolado y núcleo oval |
|
 |
Figura 3: H/E
10x: infiltración difusa por células
xantomatosas de pared colónica |
Al examinar la médula ósea, se observó una infiltración
difusa por una proliferación de estirpe linfoide de células
de apariencia linfoplasmocítica (figura 4). Las mismas eran
positivas para CD138 y CD20 con expresión de cadena ligera
kappa. El diagnóstico fue el de linfoma linfoplasmacítico
monoclonal para cadena ligera kappa sin extensión
extramedular.
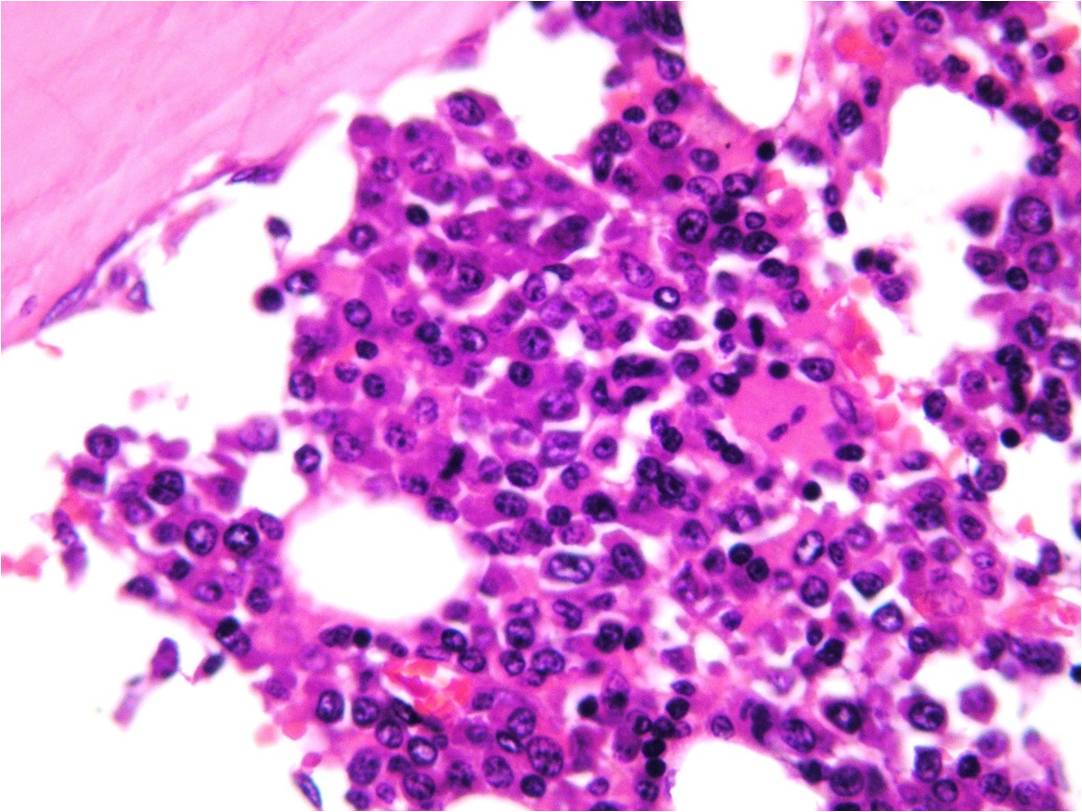 |
Figura 4: H/E 40x: infiltración de médula ósea
por células linfoides de apariencia
linfoplasmocítica |
Además, a nivel de ciego, se puso de manifiesto una lesión
tumoral ulverovegetante, friable, de 45mm de diámetro que
obstruía la luz colónica en un 70%, infiltraba la pared y se
encontraba casi en contacto con la válvula ileocecal.
Microscópicamente correspondió a un adenocarcinoma
moderadamente diferenciado con extensión a subserosa (figura
5). En los cortes seriados de hígado, se reconocieron dos
lesiones nodulares pardoblanquecinas, la mayor de 30mm,
catalogadas como metástasis de adenocarcinoma colónico.
Los eventos finales fueron: bronconeumonía aguda bilateral,
edema agudo de pulmón, síndrome de distrés respiratorio
agudo e isquemia miocárdica.
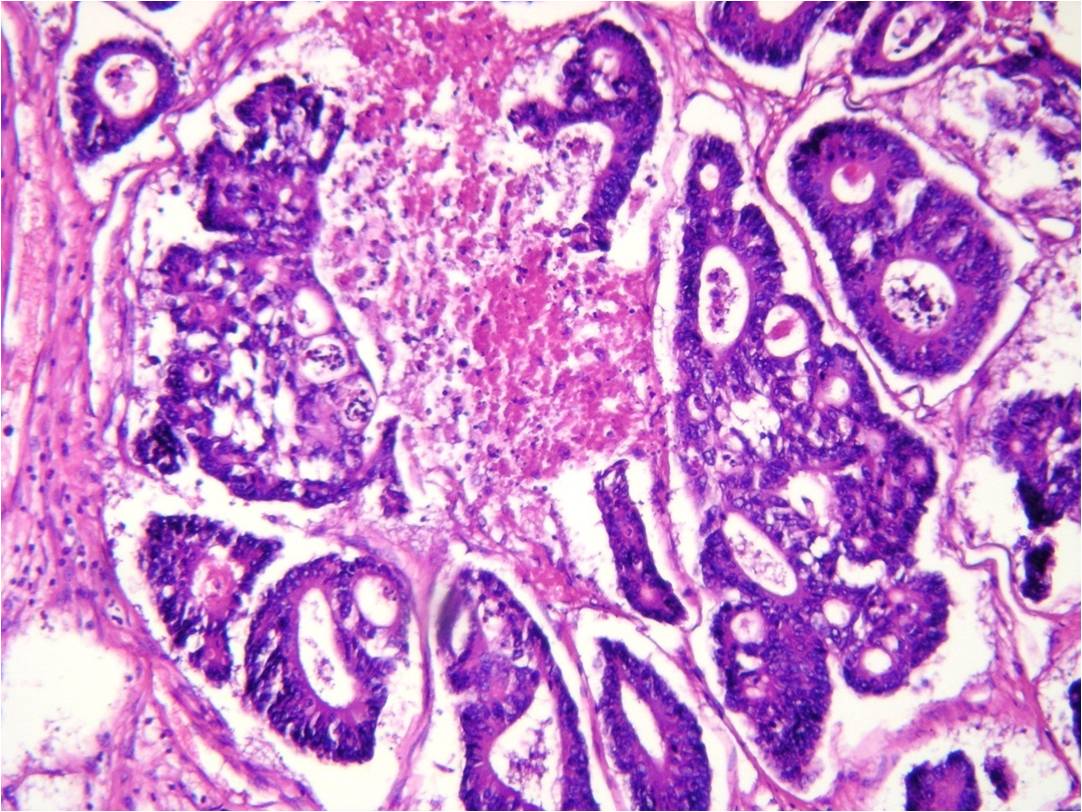 |
Figura 5: H/E
20x: infiltración de pared colónica por
adenocarcinoma moderadamente diferenciado |
Discusión
La xantomatosis plana difusa es una entidad inusual que
suele afectar la piel. Fue inicialmente descripta en el año
1962 por Altamn y Winkelmann como xantoma plano difuso
normolipémico.1
Suele presentarse en pacientes adultos en la edad media de
la vida, con una edad entre los 50 a 60 años, sin
predilección de género.,1,2
Clínicamente se caracteriza por la presencia de múltiples
máculas o pápulas pardoamarillentas diseminadas, de
crecimiento lento, que evolucionan a placas ligeramente
sobreelevadas que pueden afectar cualquier parte de la
superficie corporal, especialmente cabeza, cuello y tronco
superior. En el caso reportado, se evidencian múltiples
lesiones compatibles con predominio en dorso, cuello y
extremidades. La lesión mayor se encontraba en la región
lumbar baja y presentaba el aspecto de una gran placa
pardoamarillenta infiltrada de bordes mal definidos.1,2,3
La afectación de mucosas se ha reportado, con compromiso de
mucosa ocular, faríngea, laríngea y bronquial. Las
manifestaciones extracutáneas son menos frecuentes
compromiso de válvula aórtica, músculos y tendones, huesos,
sistema hepatobiliar, sistema nervioso central, nervios
periféricos e hipófisis. Se ha reportado un 40% de casos con
diabetes insípida debido a la acumulación de células
xantomatosas en la hipófisis. En nuestro caso se evidenció
un extenso compromiso de piel así como de tejidos internos
como serosas, cápsula hepática, tracto gastrointestinal,
corazón y mucosa vesical.4,5,6,7
Su etiopatogenia es desconocida. Una de las teorías se
refiere a la formación de un complejo lipoproteína-paraproteína
que sería fagocitada por histiocitos. Aunque en los últimos
años, se ha postulado que podría ser una xantomatosis
derivada de una histiocitosis dentro del espectro de las
histiocitosis tipo II no Langerhans, cuya célula de origen
serían células dendríticas de la dermis, clasificada
actualmente por la Histiocyte Society desde 1997.1,2,8,9
Suele tener un curso indolente y benigno, aunque se han
reportado casos con evolución tórpida dado el compromiso
sistémico.3,5,6
Se ha clasificado de la siguiente manera:
(1)
xantoma plano difuso
asociado a trastornos hematológicos, el más frecuente;
(2)
xantoma plano difuso en relación con enfermedades cuya
asociación parece meramente casual;
(3)
xantoma plano difuso que aparece tras daño de la piel;
(4)
xantoma plano difuso en relación con aumento de lípidos en
suero o secundario a cirrosis biliar o hepatopatía
obstructiva y
(5)
xantoma plano difuso esencial si no se encuentra patología
asociada.3
Al examen histopatológico suele observarse infiltración
difusa de dermis superficial y profunda por células con
núcleo oval y vesicular de citoplasma amplio y vacuolado con
disposición perivascular, perianexial y perineural, junto
con macrófagos, ocasionales células gigantes de tipo cuerpo
extraño y algunos eosinófilos. También puede observarse
depósito de hemosiderina y elastofagocitosis. La epidermis
suele estar adelgazada. Las células tumorales son positivas
para CD68 y negativas para S100 y CD1a.1,2
El principal diagnóstico diferencial es con los otros
subtipos de histiocitosis, sobre todo con la histiocitosis
de Langerhans, cuyo origen es la célula de Langerhans. Desde
el punto de vista morfológico, el patrón de compromiso de la
dermis en este tipo de histiocitosis es en banda y las
células presentan un núcleo reniforme. El perfil
inmunohistoquímico es diferente, con expresión de S100 y
CD1a.1,2,10
Su asociación con
enfermedades hematológicas fue descripta en el año 1966 por
Lynch y Winkelmann, principalmente con gammapatías
monoclonales de significado incierto y mieloma múltiple.
Existen reportes de casos en los que se ha asociado dicha
entidad con leucemia mielomonocítica crónica y leucemia
linfática crónica. Dicha asociación se presenta en el 20% de
los casos. Se ha reportado que mientras mayor número de
lesiones exista y mayor tiempo de evolución tengan las
mismas, se incrementa el riesgo de padecer una enfermedad
hematológica. En este caso, se presenta junto a un linfoma
linfoplasmacítico (LPL), que corresponde sólo al 2% de los
procesos linfoproliferativos, de estirpe B y que suele
comprometer la médula ósea con baja frecuencia de extensión
nodal o extranodal. Forma parte de las gammapatías
monoclonales ya que es frecuente la presencia de
paraproteína M con restricción de expresión de cadena
ligera. Esto es debido a la que la célula de origen
postulada sería un linfocito B post-germinal con
diferenciación a célula plasmática. Además, el 30% de los
casos se acompaña de macroglobulinemia de Waldenström y el
20%, de crioglobulinemia. Suele presentar un curso indolente
con una sobrevida de entre 5 a 10 años. En este caso, dicho
proceso linfoproliferativo impresiona tener una evolución
solapada y lenta, con confinamiento a la médula ósea. No se
puede conocer con exactitud el momento en que se produjo la
transformación neoplásica de las células B desde MGUS a LPL
durante los 16 años de evolución de la misma.3,7,11,12,13,14
Su asociación con tumores sólidos ha sido descrita con
escasa frecuencia. En 2003, Broeshart y cols. describieron
un caso que se presentó junto a un adenocarcinoma de recto.
En el presente reporte, se describe la coexistencia de un
adenocarcinoma de ciego en estadio avanzado con metástasis
hepáticas, siendo probablemente la causa desencadenante de
la muerte.15
La xantomatosis plana diseminada es una enfermedad inusual.
En este caso, se destaca el extenso compromiso de órganos
internos. Además, se debe conocer su asociación con
gammapatías monoclonales y, con menor frecuencia, con la
presencia de tumores sólidos. Por eso es importante
reconocer esta entidad y realizar un seguimiento adecuado
debido a la posibilidad de desarrollar una neoplasia
asociada.
Bibliografía
1. Calonje E, Brenn T, Lazar AJ, McKee PH. McKee´s
Pathology of the skin. 3edn. Elsevier Mosby. London.
2005:1357-1495.
2. Weedon D. Weedon’s Skin Pathology.3edn. Churchill
Livingstone/Elsevier. Philadelphia. 2010:1057-93.
3. Peón Currás, G; Fonseca Moretón, A; Veiga Codesido, C;
Gómez Domínguez, J M. Xantoma plano difuso asociado a
gammapatía monoclonal, crioglobulinemia e hipocomplementemia.
Actas Dermosifiliogr. 2001;92(3):97-100.
Full Text
4. Tietge UJ, Maschek H, Schneider A, Gawehn AE, Wagner S,
Manns MP, et al. Xanthoma disseminatum with marked
mucocutaneous involvement. Dtsch Med Wochenschr.
1998;123(45):1337-42.
5. Davies CW, Marren P, Juniper MC, Gray W, Wojnorowska F,
Benson MK. Xanthoma disseminatum with respiratory tract
involvement and fatal outcome. Thorax. 2000;55(2):170-2.
Full Text
6. Knobler RM, Neumann RA, Gebhart W, Radaskiewicz T,
Ferenci P; Widhalm K. Xanthoma disseminatum with progressive
involvement of the central nervous and hepatobiliary systems.
J Am Acad Dermatol. 1990,23:341-6.
PubMed
7. Lazrak K, Machet MC, Forest JL, Machet L, Lorette G,
Pasquiou C. Disseminated xanthosiderohistiocytosis with
cardiac involvement and monoclonal gammapathy. Ann Dermatol
Venereol. 1993;120(12):904-6.
PubMed
8. Trébol I, Acebo E, Eguino P, Gardeazabal J, Díaz-Pérez JL.
Placas amarillentas en tronco y flexuras axilares. Actas
Dermosifiliogr. 2005;96(2):124-6.
Full Text
9. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico
M. Contemporary classification of histiocytic disorders. The
WHO Committee On Histiocytic/Reticulum Cell Proliferations.
Reclassification Working Group of the Histiocyte Society.
Med Pediatr Oncol. 1997; 29(3):157-166.
PubMed
10. Zelger B, Cerio R, Orchard G, Fritsch P, Wilson-Jones E.
Histologic and immunohistochemical study comparing xanthoma
disseminatum and histiocytosis X Arch Dermatol.
1992;128:1207-12.
PubMed
11. Gómez Centeno P, García Costa A, Rodríguez López JA,
Álvarez López J, Cabo Gómez F, Kim KJ et al. Diffuse plane
xanthoma in a patient with chronic myeloid leukemia. J
Dermatol. 2004;31:503–5.
12. Maxit MJ, Paz RA. Diffuse plane xanthoma with arthritis,
serositis, erythema nodosum, vasculitis and myelomonocytic
leukemia. Description of a case with autopsy. Medicina (B
Aires). 2001;61(2):187-90.
PubMed
13. Segner S, Theate I, Poiré X, Tennstedt D, Marot L, Caers
J. Diffuse xanthomatosis as a presenting feature of multiple
myeloma. Eur J Haematol. 2010;84(5):460-1.
PubMed
14. Stockman A, Delanghe J, Geerts ML, Naeyaert JM. Diffuse
plane normolipaemic xanthomatosis in a patient with chronic
lymphatic leukaemia and monoclonal gammopathy. Dermatology.
2002;204:351–4.
PubMed
15. Broeshart JH, Prens EP, Habets WJ, de Bruijckere LM.
Normolipemic plane xanthoma associated with adenocarcinoma
and severe itch. J Am Acad Dermatol. 2003;49(1):119-22.
PubMed

|

